11 Jan 08:02
Chem. Commun., 2016, 52,2909-2912
DOI: 10.1039/C5CC09533F, Communication
Jaime Fernandez-Casado, Ronald Nelson, Jose L. Mascarenas, Fernando Lopez
Aldehydes can be [small alpha]-alkylated with allenamides by the combined action of an organocatalyst and a gold complex.
The content of this RSS Feed (c) The Royal Society of Chemistry

07 Jan 12:20
by Dean G. Brown and Jonas Boström

Journal of Medicinal Chemistry
DOI: 10.1021/acs.jmedchem.5b01409

07 Jan 12:06
by A. Stephen K. Hashmi
Abstract
We herein report a computational study of the bonding in gold(I) vinylidene complexes and compare them to their carbene and CO analogues. The relevance of these intermediates is analysed for the intramolecular cyclisation leading to vinyl sulfonates.

Tight ion pair gold vinylidene complexes are studied as intermediates in the formation of vinyl sulfonates using computational methods (see scheme). The role of π-backbonding in gold vinylidene complexes, such as the aforementioned tight ion pair, is analysed by using the intrinsic bond orbital method.
04 Jan 11:46
Chem. Sci., 2016, 7,475-481
DOI: 10.1039/C5SC03568F, Edge Article

Open Access
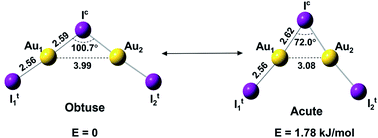
Wan-Lu Li, Hong-Tao Liu, Tian Jian, Gary V. Lopez, Zachary A. Piazza, Dao-Ling Huang, Teng-Teng Chen, Jing Su, Ping Yang, Xin Chen, Lai-Sheng Wang, Jun Li
Two isomers, different only by bond angles, are discovered for Au2I3-, due to competition between aurophilic interactions and covalent bonding.
The content of this RSS Feed (c) The Royal Society of Chemistry

04 Jan 09:22
by Laurens M. De Coen, Thomas S. A. Heugebaert, Daniel García and Christian V. Stevens

Chemical Reviews
DOI: 10.1021/acs.chemrev.5b00483

23 Dec 10:07
by Markus Warnke, Tobias Jung, Juri Dermer, Karin Hipp, Nico Jehmlich, Martin von Bergen, Sascha Ferlaino, Alexander Fries, Michael Müller, Matthias Boll
Abstract
The hydroxylation of vitamin D3 (VD3, cholecalciferol) side chains to give 25-hydroxyvitamin D3 (25OHVD3) is a crucial reaction in the formation of the circulating and biologically active forms of VD3. It is usually catalyzed by cytochrome P450 monooxygenases that depend on complex electron donor systems. Cell-free extracts and a purified Mo enzyme from a bacterium anaerobically grown with cholesterol were employed for the regioselective, ferricyanide-dependent hydroxylation of VD3 and proVD3 (7-dehydrocholesterol) into the corresponding tertiary alcohols with greater than 99 % yield. Hydroxylation of VD3 strictly depends on a cyclodextrin-assisted isomerization of VD3 into preVD3, the actual enzymatic substrate. This facile and robust method developed for 25OHVD3 synthesis is a novel example for the concept of substrate-engineered catalysis and offers an attractive alternative to chemical or O2 /electron-donor-dependent enzymatic procedures.

Well PrepareD: Oxygen-independent, regioselective, and complete hydroxylation of the tertiary C25 of vitamin D3 to its circulating 25-hydroxy form was catalyzed by a Mo-containing dehydrogenase from a denitrifying bacterium grown with cholesterol. This electron-donor-independent system can be used for the enzymatic synthesis of the clinically most relevant form of vitamin D.
23 Dec 10:06
by Michelle Francl
Nature Chemistry 8, 1 (2016).
doi:10.1038/nchem.2426
Author: Michelle Francl
Michelle Francl explores the concepts that could help non-chemists see the world more like those trained in the subject.
01 Dec 12:14
by Jose R. Cabrero-Antonino, Elisabetta Alberico, Hans-Joachim Drexler, Wolfgang Baumann, Kathrin Junge, Henrik Junge and Matthias Beller

ACS Catalysis
DOI: 10.1021/acscatal.5b01955

27 Nov 07:27
by Hongming Jin, Long Huang, Jin Xie, Matthias Rudolph, Frank Rominger, A. Stephen K. Hashmi
17 Nov 11:19
by Veerabahu Shanmugasundaram, Liying Zhang, Shilva Kayastha, Antonio de la Vega de León, Dilyana Dimova and Jürgen Bajorath

Journal of Medicinal Chemistry
DOI: 10.1021/acs.jmedchem.5b01428

17 Nov 09:37
by Justin A. Goodwin, Carl F. Ballesteros and Aaron Aponick

Organic Letters
DOI: 10.1021/acs.orglett.5b02725

17 Nov 09:30
by Alexander R. Rovira, Andrea Fin and Yitzhak Tor

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b10420

05 Nov 15:12
by Yaxi Yang, Xuan Wang, Yuanchao Li, Bing Zhou
26 Oct 12:14
by Subhasis Paul, Joyram Guin
Abstract
An operationally simple and economical method for the direct alkylation of heteroaromatic bases employing readily available aldehydes as alkyl radical precursors and molecular oxygen as a reagent is presented. This simple transformation demonstrates a broad substrate scope with respect to aldehydes and nitrogen heterocycles, enabling the introduction of several medicinally important yet challenging alkyl moieties, such as ethyl, isopropyl, tert-butyl, and cyclohexyl to the different classes of heterocyclic bases in good to excellent yields.

A simple method for the direct alkylation of heteroaromatic bases with aldehydes as inexpensive alkyl radical precursors and molecular oxygen as a reagent is presented. This transformation demonstrates a broad substrate scope with respect to aldehydes and nitrogen heterocycles, enabling the introduction of various alkyl moieties to heterocyclic bases (>40 examples) in good to excellent yields.
26 Oct 07:37
by Yi Yang See, Aaron T. Herrmann, Yoshinori Aihara and Phil S. Baran

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b09463

14 Oct 12:06
Chem. Sci., 2015, Advance Article
DOI: 10.1039/C5SC03568F, Edge Article
Open Access
Wan-Lu Li, Hong-Tao Liu, Tian Jian, Gary V. Lopez, Zachary A. Piazza, Dao-Ling Huang, Teng-Teng Chen, Jing Su, Ping Yang, Xin Chen, Lai-Sheng Wang, Jun Li
Two isomers, different only by bond angles, are discovered for Au2I3-, due to competition between aurophilic interactions and covalent bonding.
To cite this article before page numbers are assigned, use the DOI form of citation above.
The content of this RSS Feed (c) The Royal Society of Chemistry

08 Oct 09:53
Chem. Commun., 2015, 51,16806-16809
DOI: 10.1039/C5CC07018J, Communication
Marta Ayerbe Garcia, Wolfgang Frey, Mark R. Ringenberg, Max Schwilk, Rene Peters
The first scalemic ferrocenyl gold(I) and gold(II) complexes have been prepared and structurally analysed by X-ray, (spectro)electrochemical and DFT studies.
The content of this RSS Feed (c) The Royal Society of Chemistry

08 Oct 08:18
by Melissa Lee and Melanie S. Sanford

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b09099

06 Oct 12:08
by Jordi Serra, Christopher J. Whiteoak, Ferran Acuña-Parés, Marc Font, Josep M. Luis, Julio Lloret-Fillol and Xavi Ribas

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b08756

02 Oct 11:33
by Zhenxing Liu, Haocheng Tan, Tianren Fu, Ying Xia, Di Qiu, Yan Zhang and Jianbo Wang

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b09135

30 Sep 15:45
by Weiwei Zi, F. Dean Toste
Abstract
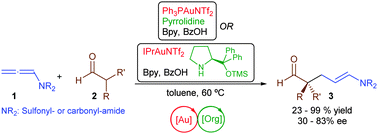
A gold(I)-catalyzed enantioselective desymmetrization of 1,3-diols was achieved by intramolecular hydroalkoxylation of allenes. The catalyst system 3-F-dppe(AuCl)2 /(R)-C8-TRIPAg proved to be specifically efficient to promote the desymmetrizing cyclization of 2-aryl-1,3-diols, which have proven challenging substrates in previous reports. Multisubstituted tetrahydrofurans were prepared in good yield with good enantioselectivity and diastereoselectivity by this method.

A chiral anion-mediated enantioselective gold(I)-catalyzed desymmetrization of 1,3-diols by intramolecular allene hydroalkoxylation was developed. Subtle tuning of the both the chiral phosphate (X*) and the achiral phosphine ligand (L) components of the catalyst system allowed for the preparation of oxygen heterocycles containing two stereocenters in high enantio- and diastereoselectivity.
15 Sep 05:55
by Jordan M. Hoyt
Cycloadditions, such as the [4+2] Diels-Alder reaction to form six-membered rings, are among the most powerful and widely used methods in synthetic chemistry. The analogous [2+2] alkene cycloaddition to synthesize cyclobutanes is kinetically accessible by photochemical methods, but the substrate scope and functional group tolerance are limited. Here, we report iron-catalyzed intermolecular [2+2] cycloaddition of unactivated alkenes and cross cycloaddition of alkenes and dienes as regio- and stereoselective routes to cyclobutanes. Through rational ligand design, development of this base metal–catalyzed method expands the chemical space accessible from abundant hydrocarbon feedstocks.
Authors: Jordan M. Hoyt, Valerie A. Schmidt, Aaron M. Tondreau, Paul J. Chirik
15 Sep 05:53
by Manoranjan Kumar, Richa, Sushila Sharma, Vinod Bhatt, Neeraj Kumar
Abstract
The first iron(III) chloride-catalyzed decarboxylative–deaminative functionalization of phenylglycine with o-substituted nitroarenes was achieved for the synthesis of 4(3H)-quinazolinones and benzimidazoles. The reaction of 2-nitrobenzonitrile/2-nitro-N,N-diphenylamine with phenylglycine at 120 °C in the presence of potassium carbonate as a base in toluene generated the products in 45–87% yields. Various functional groups like nitro, fluoride, chloride and trifluoromethyl were well tolerated under the present reaction conditions. In this tandem approach, involvement of transfer hydrogenation of the nitro functionality with in situ generated ammonia, imination, nitrile hydration to amide and oxidative cyclization sequences have been established. The process avoids the use of an external hydrogen source, costly catalysts as well as the isolation of amine and amide intermediates.

14 Aug 05:44
by Girish C. Sati and David Crich

Organic Letters
DOI: 10.1021/acs.orglett.5b02079

12 Aug 10:19
by Biao Xiong, Ya Li, Wan Lv, Zhenda Tan, Huanfeng Jiang and Min Zhang

Organic Letters
DOI: 10.1021/acs.orglett.5b01976

11 Aug 06:05
by Eugene Chong and Suzanne A. Blum

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b06678

11 Aug 05:27
by Gyorgy Szekely, Miriam C. Amores de Sousa, Marco Gil, Frederico Castelo Ferreira and William Heggie

Chemical Reviews
DOI: 10.1021/cr300095f

06 Aug 06:31
Chem. Commun., 2015, 51,12673-12676
DOI: 10.1039/C5CC03369A, Communication
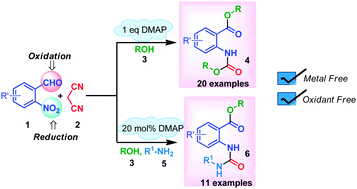
Satavisha Sarkar, Abu T. Khan
A hitherto unreported route for the synthesis of anthranilate esters is demonstrated using 2-nitrobenzaldehyde, malonitrile and an alcohol or amine via a metal and oxidant free multicomponent reaction (MCR) strategy.
The content of this RSS Feed (c) The Royal Society of Chemistry

24 Jul 10:08
by Jicheng Wu, Wenbo Xu, Zhi-Xiang Yu and Jian Wang

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b06400

24 Jul 07:48
by Line Næsborg, Kim Søholm Halskov, Fernando Tur, Sofie M. N. Mønsted, Karl Anker Jørgensen
Abstract
The first asymmetric regio- and diastereodivergent γ-allylation of cyclic α,β-unsaturated aldehydes based on combined organocatalysis and transition-metal catalysis is disclosed. By combining an aminocatalyst with an iridium catalyst, both diastereomers of branched allylated products can be achieved in moderate to good yields and excellent regio- and stereoselectivities. Furthermore, by replacing the iridium catalyst with a palladium catalyst, the linear allylated products are formed in good yields and excellent regio- and enantioselectivities. The developed method thus provides selective access to all six isomers of the γ-allylated product in a divergent fashion by choosing the appropriate combination of organocatalyst, transition-metal catalyst, and ligand.

Catalyst combo: By combining an aminocatalyst with an iridium catalyst, both diastereomers of branched allylated products can be achieved in good yields and excellent regio- and stereoselectivities. By replacing the iridium catalyst with a palladium catalyst, the linear allylated products are formed in good yields and excellent regio- and enantioselectivities. Thus, all six isomers of the γ-allylated product can be accessed.



![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) H annulation of anthranil derivatives with alkynes offers a facile, flexible, and atom-economical one-step route to unprotected 7-acylindoles. An intermediate α-imino gold carbene, generated by an intermolecular reaction, promotes ortho-aryl C
H annulation of anthranil derivatives with alkynes offers a facile, flexible, and atom-economical one-step route to unprotected 7-acylindoles. An intermediate α-imino gold carbene, generated by an intermolecular reaction, promotes ortho-aryl C