Journal of the American Chemical Society
DOI: 10.1021/jacs.7b04364


A gold(I)-catalyzed cascade [3,3]-propargylic rearrangement and [4+2] cycloaddition reaction of 2-vinylindoles with propargylic esters is reported. The reaction leads to the synthesis of highly substituted tetrahydrocarbazole derivatives in high yields and diasteroselectivities. Furthermore, a preliminary screening for an asymmetric version of this reaction is described.



An efficient entry into the phosphorylated marine macrolide enigmazole A is described. Enigmazole A interferes with c-Kit signaling by an as yet unknown mode of action and is therefore a potential lead in the quest for novel anticancer agents. Key to success is a gold-catalyzed cascade comprising a [3,3]-sigmatropic rearrangement of a propargyl acetate along the periphery of a macrocyclic scaffold, followed by a transannular hydroalkoxylation of the resulting transient allenyl acetate. This transformation mandated the use of a chiral gold catalyst to ensure a matching double-asymmetric setting. Other noteworthy steps are the preparation of the oxazole building block by a palladium-catalyzed C−H activation, as well as the smooth ring-closing alkyne metathesis of a diyne substrate bearing a propargylic leaving group, which has only little precedent.

Within reach: A gold-catalyzed cascade of remarkable complexity paved the way to the parent phosphorylated macrolide of the enigmazole family, which exhibits a highly promising but not yet well-understood biological profile. Only if the π-acid itself is chiral, however, was the [3,3]-sigmatropic rearrangement complete before the hydroxy group reached across the macrocyclic ring.


A protocol involving a gold(I)-catalyzed reaction to elaborate cyclic frames by selective trimerization reactions of N-allenylsulfonamides is reported. Efficiency in the formation of the target [2+2+2] products depends on the concentration of the reagents, the nature of the ancillary ligand on the gold catalyst, and, to a significant extent, on the selection of a low reaction temperature. This singular catalytic allene cycloaddition gives adducts in high isolated yields. In addition to representing a new gold-catalyzed reaction, the reported allenamide cyclotrimerization offers a significant extension to the scant number of allenes that have effectively entered this cyclization mode by use of other metal-catalyzed reactions. Upon aromatization of the assembled adducts, the overall reaction matches the outcome of related and widely investigated alkyne trimerization reactions.

Granting gilding access to triskelium-like molecules: The first examples of a gold-catalyzed [2+2+2]-cycloaddition reaction of tosylallenamides are presented. Upon ligand screening, conditions have been devised to favor the direct assembly of molecules with this particular shape.
3-Oxo-5-alkynoic acid esters, on treatment with a carbophilic catalyst, undergo 6-endo-dig cyclization reactions to furnish either 2-pyrones or 4-pyrones in high yields. The regiochemical course can be dialed in by the proper choice of the alcohol part of the ester and the π-acid. This transformation is compatible with a variety of acid-sensitive groups as witnessed by a number of exigent applications to the total synthesis of natural products, including pseudopyronine A, hispidine, phellinin A, the radininol family, neurymenolide, violapyrone, wailupemycin and an unnamed brominated 4-pyrone of marine origin. Although the reaction proceeds well in neutral medium, the rate is largely increased when HOAc is used as solvent or co-solvent, which is thought to favor the protodeauration of the reactive alkenyl-gold intermediates as the likely rate-determining step of the catalytic cycle. Such intermediates are prone to undergo diauration as an off-cycle event that sequesters the catalyst; this notion is consistent with literature data and supported by the isolation of the gem-diaurated complexes 12 and 15. Furthermore, silver catalysis allowed access to be gained to 2-alkoxypyridine and 2-alkoxyisoquinoline derivatives starting from readily available imidate precursors.

Dial-in: Upon proper choice of the ester terminus and the carbophilic catalyst, readily accessible 3-oxo-5-ynoate derivatives are converted either into 4-hydroxy-2-pyrones or 2-alkoxy-4-pyrones, as one desires. The conditions are unusually mild, as witnessed by a number of applications to delicate natural products. The underlying concept has also been extended to the preparation of N-heterocycles such as 2-alkoxypyridines or -isoquinolines.
 Open Access
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.

Open Access  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
Open Access  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
The mechanism of OsH6(PiPr3)2-mediated fragmentation of a 4-(2 pyridyl)-2-azetidinone has been investigated by DFT calculations. The addition of the C4![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) H bond of the substrate to OsH2(PiPr3)2 allows the active participation of an osmium lone pair in the B-type β-lactam fragmentation process. This new mechanism makes the N1C4/C2C3 fragmentation of the lactamic core thermally accessible through a stepwise process.
H bond of the substrate to OsH2(PiPr3)2 allows the active participation of an osmium lone pair in the B-type β-lactam fragmentation process. This new mechanism makes the N1C4/C2C3 fragmentation of the lactamic core thermally accessible through a stepwise process.

Wizard of Os: Based on DFT calculations, the complex OsH6(PiPr3)2 breaks the lactamic core of 4-(2-pyridyl)-2-azetidinones through addition of the C4H bond to the metal center by means of a new mechanism entailing the active participation of an osmium lone pair.
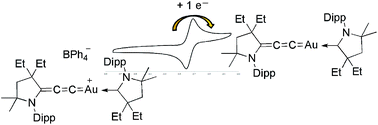
Reactions of the gold(I) triflimide complex [Au(NTf2)(PMe2Ar )] (1) with the gold(I) hydrocarbyl species [AuR(PMe2Ar
)] (1) with the gold(I) hydrocarbyl species [AuR(PMe2Ar )] (2 a–2 c) enable the isolation of hydrocarbyl-bridged cationic digold complexes with the general composition [Au2(μ-R)(PMe2Ar
)] (2 a–2 c) enable the isolation of hydrocarbyl-bridged cationic digold complexes with the general composition [Au2(μ-R)(PMe2Ar )2][NTf2], where Ar
)2][NTf2], where Ar =C6H3-2,6-(C6H3-2,6-iPr2)2 and R=Me (3), CH
=C6H3-2,6-(C6H3-2,6-iPr2)2 and R=Me (3), CH![[DOUBLE BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8fe.gif) CH2 (4), or C
CH2 (4), or C![[TRIPLE BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8fd.gif) CH (5). Compound 3 is the first alkyl-bridged digold complex to be reported and features a symmetric [Au(μ-CH3)Au]+ core. Complexes 4 and 5 are the first species of their kind that contain simple, unsubstituted vinyl and acetylide units, respectively. In the series of complexes 3–5, the bridging carbon atom systematically changes its hybridization from sp3 to sp2 and sp. Concomitant with this change, and owing to variations in the nature of the bonding within the [Au(μ-R)Au]+ unit, there is a gradual decrease in aurophilicity, that is, the strength of the Au⋅⋅⋅Au bonding interaction decreases. This change is illustrated by a monotonic increase in the Au–Au distance by approximately 0.3 Å from R=CH3 (2.71 Å) to CHCH2 (3.07 Å) and CCH (3.31 Å).
CH (5). Compound 3 is the first alkyl-bridged digold complex to be reported and features a symmetric [Au(μ-CH3)Au]+ core. Complexes 4 and 5 are the first species of their kind that contain simple, unsubstituted vinyl and acetylide units, respectively. In the series of complexes 3–5, the bridging carbon atom systematically changes its hybridization from sp3 to sp2 and sp. Concomitant with this change, and owing to variations in the nature of the bonding within the [Au(μ-R)Au]+ unit, there is a gradual decrease in aurophilicity, that is, the strength of the Au⋅⋅⋅Au bonding interaction decreases. This change is illustrated by a monotonic increase in the Au–Au distance by approximately 0.3 Å from R=CH3 (2.71 Å) to CHCH2 (3.07 Å) and CCH (3.31 Å).

The first methyl-bridged cationic digold complex, [(
2,6-iPr2)2), is stabilized by a bulky terphenylphosphine ligand. The aurophilic interaction in this complex is comparable to that in {Au2(μ-H)}+ species and stronger than in the vinyl- and acetylide-bridged analogues (see scheme).
Open Access  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
An enantio-, diastereo-, regio-, and chemoselective phosphine-catalyzed β,γ-umpolung domino reaction of allenic esters with dienones has been developed for the first time. The designed sequence, involving oxy-Michael and Rauhut–Currier reactions, produced highly functionalized tetrahydrobenzofuranones, bearing a chiral tetrasubstituted stereogenic center, in up to 96 % ee.

Domino effect: An enantio-, diastereo-, regio-, and chemoselective phosphine-catalyzed β,γ-umpolung domino reaction of allenic esters with dienones has been developed for the first time. The designed sequence involving oxy-Michael and Rauhut–Currier reactions produced highly functionalized tetrahydrobenzofuranones bearing a chiral tetrasubstituted stereogenic center in up to 96 % ee.
Cyclic alkenyl triflates are useful intermediates in organic synthesis usually synthesized from ketones through a reaction involving enolization and trapping with a triflating agent. This sequence suffers from some stereochemical drawbacks owing to the basic conditions required. Herein, we describe a new acid-mediated cationic cyclization reaction of enyne derivatives (or alkynols) to access cyclic alkenyl triflates. This new atom-economical process is high yielding, scalable, technically very simple, proceeds without the need of any metallic reagent or catalyst, and more importantly, it complements and challenges conventional methodologies. We have also developed new biomimetic cationic cyclization reactions to yield interesting polycyclic compounds. As a demonstration of the potential of this method in the context of total synthesis, we have synthesized two terpenes: austrodoral and pallescensin A. Using the cationic cyclization in the key step of the synthetic routes allowed the synthesis of these natural products in a very simple, concise, scalable, and efficient way.

Cyclic alkenyl triflates are easily available through a new reaction based on a cationic cyclization process. Extension of the method to biomimetic polycyclization reactions allows the selective synthesis of interesting polycyclic core skeletons, including the terpenes austrodoral and pallescensin A.
The gold-catalyzed synthesis of methylidene 2,3-cyclobutane-indoles is documented through a combined experimental/computational investigation. Besides optimizing the racemic synthesis of the tricyclic indole compounds, the enantioselective variant is presented to its full extent. In particular, the scope of the reaction encompasses both aryloxyallenes and allenamides as electrophilic partners providing high yields and excellent stereochemical controls in the desired cycloadducts. The computational (DFT) investigation has fully elucidated the reaction mechanism providing clear evidence for a two-step reaction. Two parallel reaction pathways explain the regioisomeric products obtained under kinetic and thermodynamic conditions. In both cases, the dearomative CC bond-forming event turned out to be the rate-determining step.

2+2=3: The formal gold-catalyzed [2+2]-cycloaddition between indoles and electron-rich allenes (i.e., allenamides, alkoxyallenes) is described, providing access to substituted tricyclic cyclobutyl-indolines in high yields (see scheme). Mechanistic insights and enantioselective variants are also documented with a combined experimental/computational investigation.
Belén RubialNo tenemos acceso pero los dibujos monísimos ¬¬

Nature Chemistry. doi:10.1038/nchem.2367
Authors: Jonas Calleja, Daniel Pla, Timothy W. Gorman, Victoriano Domingo, Benjamin Haffemayer & Matthew J. Gaunt
The functionalization of primary amines by C–H activation is often hindered by their strong metal-coordinating properties. Now, a steric tethering approach — which temporarily converts amino alcohols into hindered secondary amines — has been developed. The approach allows these amino alcohols to be transformed into structurally complex and diverse products using palladium-catalysed aliphatic C–H activation.
Belén RubialYo quiero trabajar en esto! :DDD
This paper presents firm evidence for the chemical alteration of chrome yellow pigments in Van Gogh’s Sunflowers (Van Gogh Museum, Amsterdam). Noninvasive in situ spectroscopic analysis at several spots on the painting, combined with synchrotron-radiation-based X-ray investigations of two microsamples, revealed the presence of different types of chrome yellow used by Van Gogh, including the lightfast PbCrO4 and the sulfur-rich PbCr1−xSxO4 (x≈0.5) variety that is known for its high propensity to undergo photoinduced reduction. The products of this degradation process, i.e., CrIII compounds, were found at the interface between the paint and the varnish. Selected locations of the painting with the highest risk of color modification by chemical deterioration of chrome yellow are identified, thus calling for careful monitoring in the future.

Why is the yellow darkening? Firm evidence that chrome yellow pigments darken through the CrVI![[RIGHTWARDS ARROW]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/002192.gif) CrIII reduction is provided by the use of complementary analytical techniques. Different types of chrome yellow, that is, the lightfast PbCrO4 and the light-sensitive sulfur-rich PbCr1−xSxO4 (x≈0.5), are shown to be present, and spots of the painting with the highest risk of color change have been identified.
CrIII reduction is provided by the use of complementary analytical techniques. Different types of chrome yellow, that is, the lightfast PbCrO4 and the light-sensitive sulfur-rich PbCr1−xSxO4 (x≈0.5), are shown to be present, and spots of the painting with the highest risk of color change have been identified.
A mild and efficient gold-catalyzed oxidative ring-expansion of a series of alkynyl heterocycles using pyridine-N-oxide as the oxidant has been developed, which affords highly valuable six- or seven-membered heterocycles with wide functional group toleration. The reaction consists of a regioselective oxidation and a chemoselective migration of an endocyclic carbon–heteroatom bond (favored over CH migration) with the order of migratory aptitude for carbon–heteroatom bonds being CS>CN>CO. In the absence of an oxidant, polycyclic products are readily constructed through a ring-expansion/Nazarov cyclization reaction sequence.

Bigger rings through migration: A mild and efficient gold-catalyzed oxidative ring-expansion of a series of alkynyl heterocycles has been developed. The reaction features a regioselective oxidation and chemoselective migration of an endocyclic CX bond. In the absence of an oxidant, polycyclic products are readily constructed through a ring-expansion/Nazarov cyclization reaction sequence.
The activity of HBF4 (aqueous solution) as a catalyst in propargylation reactions is presented. Diverse types of nucleophiles were employed in order to form new C–O, C–N and C–C bonds in technical acetone and in air. Good to excellent yields and good chemoselectivities were obtained using low acid loading (typically 1 mol-%) under simple reaction conditions.

The activity of HBF4 (aq. solution) as a catalyst in propargylation reactions is presented. C–O, C–N and C–C bonds were formed in technical acetone and in air. Good to excellent yields were obtained using low acid loading (typically 1 mol-%) under mild reaction conditions.