Why it is not a ‘failure’ to leave academia
Why it is not a ‘failure’ to leave academia, Published online: 01 August 2018; doi:10.1038/d41586-018-05838-y
Here’s how PhD students can prepare for different careers, and how lab heads can help.Why it is not a ‘failure’ to leave academia
Why it is not a ‘failure’ to leave academia, Published online: 01 August 2018; doi:10.1038/d41586-018-05838-y
Here’s how PhD students can prepare for different careers, and how lab heads can help.Direct functionalization of natural products is important for studying the structure–activity and structure–property relationships of these molecules. Recent advances in the transition-metal-catalyzed functionalization of C(sp3)−H bonds, the most abundant yet inert bonds in natural products, have allowed natural product derivatives to be created selectively. Strategies to achieve such transformation are reviewed.

Direct derivatization: Direct functionalization of natural products is important for studying their structure–activity and structure–property relationships. Recent advances in the transition-metal-catalyzed functionalization of C(sp3)−H bonds have shown that natural product derivatives can be selectively produced. Strategies for achieving such transformations are discussed.

Small-molecule dual hydrogen-bond (H-bond) donors such as ureas, thioureas, squaramides, and guanidinium ions enjoy widespread use as effective catalysts for promoting a variety of enantioselective reactions. However, these catalysts are only weakly acidic and therefore require highly reactive electrophilic substrates to be effective. We introduce here a mode of catalytic activity with chiral H-bond donors that enables enantioselective reactions of relatively unreactive electrophiles. Squaramides are shown to interact with silyl triflates by binding the triflate counterion to form a stable, yet highly Lewis acidic, complex. The silyl triflate-chiral squaramide combination promotes the generation of oxocarbenium intermediates from acetal substrates at low temperatures. Enantioselectivity in nucleophile additions to the cationic intermediates is then controlled through a network of noncovalent interactions between the squaramide catalyst and the oxocarbenium triflate.
The development of base metal catalysts for the synthesis of pharmaceutically relevant compounds remains an important goal of chemical research. Here, we report that cobalt nanoparticles encapsulated by a graphitic shell are broadly effective reductive amination catalysts. Their convenient and practical preparation entailed template assembly of cobalt-diamine-dicarboxylic acid metal organic frameworks on carbon and subsequent pyrolysis under inert atmosphere. The resulting stable and reusable catalysts were active for synthesis of primary, secondary, tertiary, and N-methylamines (more than 140 examples). The reaction couples easily accessible carbonyl compounds (aldehydes and ketones) with ammonia, amines, or nitro compounds, and molecular hydrogen under industrially viable and scalable conditions, offering cost-effective access to numerous amines, amino acid derivatives, and more complex drug targets.

“… Drug-delivery research is experiencing a major expansion, however the upsurge in published reports does not correlate with therapeutic advances. Reporting complex systems seems to have prevailed over the desire to treat a disease effectively with a robust and safe formulation. Another important issue is the lack of reproducibility of published findings …” Read more in the Editorial by Jean-Christoph Leroux.
Enantiopure (2R)-configured non-natural (−)-muscarine and (+)-allo-muscarine were efficiently synthesized by a chemoenzymatic approach from an easily accessible cyano-sugar available on a large-scale. The key enzymatic hydrolysis step was accomplished by immobilized Burkholderia cepacia lipase (also known as Pseudomonas cepacia lipase, PSL-IM). The PSL-IM-mediated regioselective hydrolysis of the 5-O-toluoyl ester group in α- and β-cyano-sugars, furnished 3-O-toluoyl-α-cyano-sugar and 3-O-toluoyl-β-cyano-sugar, respectively, with total selectivity and in excellent yields. To demonstrate the industrial utility of this method, 3-O-toluoyl-β-cyano-sugar was synthesized on a 10 g scale using PSL-IM and the catalyst was reused. A key feature of the synthesis route described herein is the simultaneous hydrogenolysis of a tosyl group, reduction of a nitrile and deprotection of a toluoyl group using LiAlH4 in a one-pot procedure. This study offers a green protocol for synthesis of two muscarine derivatives in high yields employing a concise four-step strategy.


José Barluenga, emeritus professor at the University of Oviedo, passed away on September 7, 2016. Barluenga was one of the leading Spanish chemists of his generation, with research interests including metal-mediated activation of alkenes and alkynes, β-functionalized organolithium compounds, iodination reactions, asymmetric Diels–Alder reactions, carbene complexes, and cross-coupling reactions.

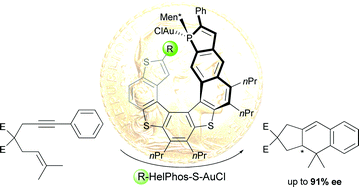
The first enantioselective intramolecular C−H insertion and cyclopropanation reactions of donor- and donor/donor-carbenes by a nondiazo approach are reported. The reactions were conducted in a one-pot manner without slow addition and provided the desired dihydroindole, dihydrobenzofuran, tetrahydrofuran, and tetrahydropyrrole derivatives with up to 99 % ee and 100 % atom efficiency.

Generous donor: Enantioselective intramolecular C−H insertion and cyclopropanation reactions of donor- and donor/donor-carbenes by a nondiazo approach are reported. The reactions were conducted in a one-pot manner without slow addition and provided the desired dihydroindole, dihydrobenzofuran, tetrahydrofuran, and tetrahydropyrrole derivatives with up to 99 % ee.
Carbene transition-metal complexes have become a prevalent family of catalysts enabling numerous organic transformations. Their facile synthetic access is a matter of great importance. To this end, the CuI-NHC transfer methodology has emerged as a powerful alternative presenting attractive advantages over other methods. Herein, we report the remarkable ability of copper to transfer not only NHCs but also other types of carbenes such as abnormal NHCs (aNHCs), cyclic (alkyl)(amino)carbenes (CAACs), and mesoionic carbenes (MICs) to various transition metal precursors.

Copper to carbene made easy! The NHC-CuI transmetallation reaction is expanded to Group 9 metals, and generalized to other carbenes such as MICs, aNHCs, and CAACs to generate a library of transition-metal complexes. Since all copper-complex precursors are made by direct metallation of easy-to-handle carbene conjugate acids with copper oxide, the methodology reported here allows for the preparation of a variety of carbene complexes without the use of highly sensitive reagents.
The observation and investigation of nonlinear effects in catalytic reactions provides valuable mechanistic insight. However, the applicability of this method was, until now, limited to molecules possessing chirality and therefore to asymmetric synthesis. The concept of nonlinear effects is expanded to catalytic reactions beyond asymmetric catalysis by using derivatives instead of enantiomers and by considering rates instead of enantiomeric excess. Additionally, its systematic application to investigate the mechanism of catalytic reactions is presented. By exceeding the limitation to asymmetric reactions, the study of nonlinear effects can become a general tool to elucidate reaction mechanisms.

Escaping asymmetry: The concept of nonlinear effects is expanded from asymmetric catalysis to catalytic reactions in general by investigating reaction rates instead of enantiomeric purity and using derivatives instead of enantiomers.




The use of micrometric hollow silica spheres is described as a strategy to reduce magnetic field inhomogeneities in the context of NMR chromatography. When employed as a stationary phase, hollow silica microspheres allow the use of common solution-state NMR instruments to measure the diffusion coefficient perturbation induced by the interaction of the analytes with the silica surface.

A (w)hole lot better: Hollow silica microspheres made possible the use of solution-state NMR instruments to analyze mixtures of small molecules by chromatographic NMR spectroscopy (see picture). The silica microspheres clearly provide major advantages over conventional stationary phases.
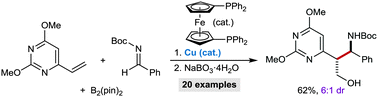
Hydrogenation of esters to alcohols with a well-defined iron iPr2PNP pincer complex has been recently reported by us and other groups. We now introduce a novel and sterically less hindered Et2PNP congener that provides superior catalytic activity in the hydrogenation of various carboxylic acid esters and lactones compared to the known complex. Successful hydrogenation proceeds under relatively mild conditions (60 °C) with lower catalyst loadings.

Terminal alkynes (RCCH) are homologated by a sequence of ruthenium-catalyzed anti-Markovnikov hydration of alkyne to aldehyde (RCH2CHO), followed by Bestmann–Ohira alkynylation of aldehyde to chain-elongated alkyne (RCH2CCH). Inverting the sequence by starting from aldehyde brings about the reciprocal homologation of aldehydes instead. The use of 13C-labeled Bestmann–Ohira reagent (dimethyl ((1-13C)-1-diazo-2-oxopropyl)phosphonate) for alkynylation provides straightforward access to singly or, through additional homologation, multiply 13C-labeled alkynes. The labeled alkynes serve as synthetic platform for accessing a multitude of specifically 13C-labeled products. Terminal alkynes with one or two 13C-labels in the alkyne unit have been submitted to alkyne–azide click reactions; the copper-catalyzed version (CuAAC) was found to display a regioselectivity of >50 000:1 for the 1,4- over the 1,5-triazine isomer, as shown analytically by 13C NMR spectroscopy.

Really Stretching it: “True” homologations for either terminal alkynes or aldehydes are provided by a sequence of ruthenium-catalyzed anti-Markovnikov hydration of terminal alkynes to aldehydes, followed by Bestmann–Ohira alkynylation of aldehydes to alkynes (see scheme). The reaction opens a general access to 13C-labeled linear targets, which, in the case of alkynes, can serve as starting materials for alkyne–azide “13Click” reactions.
 Open Access
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
A highly selective reductive amination of ketones to primary amines with ammonia and hydrogen using a simple ruthenium catalyst has been developed. The protocol described constitutes an efficient and direct atom-economical approach en route to α-methylbenzylamine derivatives in good to high yields. The presence of catalytic amounts of aluminum triflate turned out to be crucial for achieving high conversion towards primary amines.

Direct arylation of cyclopentanones has been a long-standing challenge because of competitive self-aldol condensation and multiple arylations. Reported herein is a direct mono-α-C![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) H arylation of cyclopentanones with aryl bromides which is enabled by palladium/amine cooperative catalysis. This method is scalable and chemoselective with broad functional-group tolerance. Application to controlled sequential arylation of cyclopentanones has been also demonstrated.
H arylation of cyclopentanones with aryl bromides which is enabled by palladium/amine cooperative catalysis. This method is scalable and chemoselective with broad functional-group tolerance. Application to controlled sequential arylation of cyclopentanones has been also demonstrated.

Be direct: A direct α-CH arylation of normal cyclopentanones with aryl bromides, enabled by palladium/amine cooperative catalysis, features an exceptionally high selectivity for monoarylation, use of readily available starting materials, good scalability, and broad functional-group tolerance.