14 Jul 13:44
by Amy J. Challinor, Marc Calin, Gary S. Nichol, Neil B. Carter, Stephen P. Thomas
Abstract
A simple alkylamine [(iPr)2NEt] has been used to activate an air- and moisture-stable iron(II) pre-catalyst for alkene and alkyne hydrofunctionalization reactions. This amine activation has enabled the highly operationally simple hydrosilylation and hydroboration of alkenes and alkynes using just 0.25–2 mol% iron catalyst and 1–25 mol% amine. Significantly, these reactions proceed in equal yield under both air and inert reaction conditions.

14 Jul 13:43
by Pierre-Georges Echeverria, Alois Fürstner
Abstract
Treatment of readily available enynes with alkyl-Grignard reagents in the presence of catalytic amounts of Fe(acac)3 engenders a remarkably facile and efficient reaction cascade that results in the net formation of two new C−C bonds while a C−Z bond in the substrate backbone is broken. Not only does this new manifold lend itself to the extrusion of heteroelements (Z=O, NR), but it can even be used for the cleavage of activated C−C bonds. The reaction likely proceeds via metallacyclic intermediates, the iron center of which gains ate character before reductive elimination occurs. The overall transformation represents a previously unknown merger of cycloisomerization and cross-coupling chemistry. It provides ready access to highly functionalized 1,3-dienes comprising a stereodefined tetrasubstituted alkene unit, which are difficult to make by conventional means.

Cut and paste: Iron catalysis allows cycloisomerization chemistry and cross-coupling to be merged into a new reaction cascade; this manifold engenders formation of two new C−C bonds at the expense of a C−Z bond in the substrate backbone, which is cleaved while the tetrasubstituted alkene product is forming.
14 Jul 10:48
by Tesia V. Chciuk, William R. Anderson and Robert A. Flowers

Journal of the American Chemical Society
DOI: 10.1021/jacs.6b05879

07 Jul 16:01
by Jie Wang, Tian Qin, Tie-Gen Chen, Laurin Wimmer, Jacob T. Edwards, Josep Cornella, Benjamin Vokits, Scott A. Shaw, Phil S. Baran
Abstract
A transformation analogous in simplicity and functional group tolerance to the venerable Suzuki cross-coupling between alkyl-carboxylic acids and boronic acids is described. This Ni-catalyzed reaction relies upon the activation of alkyl carboxylic acids as their redox-active ester derivatives, specifically N-hydroxy-tetrachlorophthalimide (TCNHPI), and proceeds in a practical and scalable fashion. The inexpensive nature of the reaction components (NiCl2⋅6 H2O—$9.5 mol−1, Et3N) coupled to the virtually unlimited commercial catalog of available starting materials bodes well for its rapid adoption.

A cross-coupling between alkyl carboxylic acids and aryl boronic acids is enabled by a new activation/cross-coupling strategy under Nickel catalysis. The operational simplicity and the wide range of heterocyclic compounds make a convenient strategy for obtaining aryl–alkyl cross-coupling products.
07 Jul 15:52
Chem. Commun., 2016, 52,10817-10829
DOI: 10.1039/C6CC04359C, Feature Article

Open Access
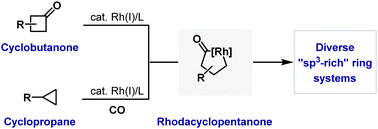
Megan H. Shaw, John F. Bower
Methodologies that exploit rhodacyclopentanones derived from C-C bond activation are outlined.
The content of this RSS Feed (c) The Royal Society of Chemistry

07 Jul 15:46
by Aymeric P. Colleville, Richard A. J. Horan, Sandrine Olazabal and Nicholas C. O. Tomkinson

Organic Process Research & Development
DOI: 10.1021/acs.oprd.6b00117

07 Jul 12:52
by Daniel Whitaker, Jordi Burés and Igor Larrosa

Journal of the American Chemical Society
DOI: 10.1021/jacs.6b04726

04 Jul 18:24
by Julian G. West, T. Aaron Bedell, Erik J. Sorensen
Abstract
The fluorination of unactivated C(sp3)−H bonds remains a desirable and challenging transformation for pharmaceutical, agricultural, and materials scientists. Previous methods for this transformation have used bench-stable fluorine atom sources; however, many still rely on the use of UV-active photocatalysts for the requisite high-energy hydrogen atom abstraction event. Uranyl nitrate hexahydrate is described as a convenient, hydrogen atom abstraction catalyst that can mediate fluorinations of certain alkanes upon activation with visible light.

U can do it: The uranyl cation (UO22+) is able to effect the catalytic fluorination of unactivated C(sp3)−H bonds under visible-light irradiation. Uranyl nitrate is highlighted as a convenient molecular C−H abstraction catalyst, which exhibits selectivity that is distinct from previously reported catalytic systems. NFSI: N-fluorobenzenesulfonimide.
04 Jul 18:15
by Wangqing Kong, Qian Wang, Jieping Zhu
Abstract
Palladium-catalyzed intramolecular carbopalladation of N-aryl acrylamides followed by migratory insertion of an isocyanide-coordinated C(sp3)−Pd intermediate afforded an alkylimidoyl−PdII complex, which can be intercepted by a nucleophile, including heteroarenes. In addition to amides, the alkylimidoyl−PdII complex was successfully converted into esters, ketones, and bis-heterocyclic compounds. An unprecedented palladium-catalyzed enantioselective domino process involving isocyanide was also documented.

Setting a trap: The palladium-catalyzed reaction of N-aryl acrylamides with isocyanides affords diversely functionalized 3,3-disubstituted oxindoles by carbopalladation, migratory insertion of isocyanide, and nucleophilic trapping of the resulting alkylimidoyl–PdII complex. An enantioselective protocol has also been developed using 2,6-diisopropylphenyl isocyanide as a reaction partner and a chiral ligand.
04 Jul 17:40
by Monica D. Lotz, Nicole M. Camasso, Allan J. Canty and Melanie S. Sanford

Organometallics
DOI: 10.1021/acs.organomet.6b00437

13 Jun 14:09
by Monya Baker
The truth about reproducibility
Nature 533, 7604 (2016). http://www.nature.com/doifinder/10.1038/533452a
Author: Monya Baker
1,500 scientists say what they really think about science’s looming ‘crisis’.

09 Jun 14:13
by Salvador B. Muñoz III, Stephanie L. Daifuku, William W. Brennessel and Michael L. Neidig

Journal of the American Chemical Society
DOI: 10.1021/jacs.6b03760

13 May 09:11
by Sugiyama, Yu-ki
Synthesis
DOI: 10.1055/s-0035-1561627

A novel process for the reductive magnesiation of 2-substituted oxetanes and the metalative cyclization of ω-alkynyl oxetanes is developed using n-propylmagnesium chloride in the presence of an iron catalyst. The generated intermediate organomagnesium compounds react with electrophiles. The reactions of 2,3-disubstituted oxetanes and their subsequent allylation with allyl halides in the presence or absence of copper(I) cyanide as the catalyst is studied with a unique switching of stereoselectivity being observed in the absence or presence of copper(I) cyanide . In addition, it is found that the metalative cyclization of 3-substituted 2-alkynyl oxetanes proceeds in an anti-selective manner starting from both syn- and anti-substrates. In all cases, the stereochemistry at the 2-position of the oxetanes is lost during the reactions suggesting the involvement of a radical process.
[...]
© Georg Thieme Verlag Stuttgart · New York
Article in Thieme eJournals:
Table of contents | Abstract | Full text
13 May 08:42
by Weiping Liu and Lutz Ackermann

ACS Catalysis
DOI: 10.1021/acscatal.6b00993

08 May 10:26
by Steven P. Cummings, Thanh-Ngoc Le, Gilberto E. Fernandez, Lorenzo G. Quiambao and Benjamin J. Stokes

Journal of the American Chemical Society
DOI: 10.1021/jacs.6b02132

海滨稚子, XJ and 4 others like this
05 May 12:30
by Ana Cristina Parra Rivera, Raymond Still, Doug E. Frantz
Abstract
A practical and highly stereoselective iron-catalyzed cross-coupling reaction of stereodefined enol carbamates and Grignard reagents to yield tri- and tetrasubstituted acrylates is reported. A facile method for the stereoselective generation of these enol carbamates has also been developed.

Ironing out olefin synthesis: A practical and highly stereoselective iron-catalyzed cross-coupling reaction between stereodefined enol carbamates and alkyl Grignard reagents yields tri- and tetrasubstituted acrylates. A facile method for the stereoselective generation of these enol carbamates is also reported.
05 May 12:30
by Ana Conde, Gerard Sabenya, Mònica Rodríguez, Verònica Postils, Josep M. Luis, M. Mar Díaz-Requejo, Miquel Costas, Pedro J. Pérez
Abstract
The first examples of the direct functionalization of non-activated aryl sp2 C−H bonds with ethyl diazoacetate (N2CHCO2Et) catalyzed by Mn- or Fe-based complexes in a completely selective manner are reported, with no formation of the frequently observed cycloheptatriene derivatives through competing Buchner reaction. The best catalysts are FeII or MnII complexes bearing the tetradentate pytacn ligand (pytacn= 1-(2-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane). When using alkylbenzenes, the alkylic C(sp3)−H bonds of the substituents remained unmodified, thus the reaction being also selective toward functionalization of sp2 C−H bonds.

Exclusive catalysis: Iron- and-manganese-based catalysts selectively functionalize the C(sp2)−H bonds of benzene or alkylbenzenes through the formal insertion of the CHCO2Et group from N2CHCO2Et (see scheme). When using alkylbenzenes, the alkylic C(sp3)−H bonds of the substituents remain unmodified.
05 May 12:11
by Jacob R. Ludwig
The olefin metathesis reaction of two unsaturated substrates is one of the most powerful carbon–carbon-bond-forming reactions in organic chemistry. Specifically, the catalytic olefin metathesis reaction has led to profound developments in the synthesis of molecules relevant to the petroleum, materials, agricultural and pharmaceutical industries. These reactions are characterized by their use of discrete metal alkylidene catalysts that operate via a well-established mechanism. While the corresponding carbonyl–olefin metathesis reaction can also be used to construct carbon–carbon bonds, currently available methods are scarce and severely hampered by either harsh reaction conditions or the required use of stoichiometric transition metals as reagents. To date, no general protocol for catalytic carbonyl–olefin metathesis has been reported. Here we demonstrate a catalytic carbonyl–olefin ring-closing metathesis reaction that uses iron, an Earth-abundant and environmentally benign transition metal, as a catalyst. This transformation accommodates a variety of substrates and is distinguished by its operational simplicity, mild reaction conditions, high functional-group tolerance, and amenability to gram-scale synthesis. We anticipate that these characteristics, coupled with the efficiency of this reaction, will allow for further advances in areas that have historically been enhanced by olefin metathesis.
Nature doi: 10.1038/nature17432

25 Apr 13:21
by Manjeet K. Majhail, Paul M. Ylioja, Michael C. Willis
Abstract
Rhodium(I) catalysts incorporating small bite-angle diphosphine ligands, such as (Cy2P)2NMe or bis(diphenylphosphino)methane (dppm), are effective at catalysing the union of aldehydes and propargylic amines to deliver the linear hydroacylation adducts in good yields and with high selectivities. In situ treatment of the hydroacylation adducts with p-TSA triggers a dehydrative cyclisation to provide the corresponding pyrroles. The use of allylic amines, in place of the propargylic substrates, delivers functionalised dihydropyrroles. The hydroacylation reactions can also be combined in a cascade process with a RhI-catalysed Suzuki-type coupling employing aryl boronic acids, providing a three-component assembly of highly substituted pyrroles.

Down the line: Rhodium catalysts featuring small-bite-angle bisphosphine ligands allow the linear-selective combination of aldehydes and propargylic amines (see scheme). The resultant γ-amino-enone products are converted in situ to a diverse range of substituted pyrroles. Allylic amine substrates can also be employed, leading in these cases to dihydropyrrole products.
25 Apr 13:20
by Davide Ravelli, Stefano Protti and Maurizio Fagnoni

Chemical Reviews
DOI: 10.1021/acs.chemrev.5b00662

25 Apr 13:19
by Jay J. Dunsford, David J. Evans, Thomas Pugh, Sachin N. Shah, Nicholas F. Chilton and Michael J. Ingleson

Organometallics
DOI: 10.1021/acs.organomet.6b00121

25 Apr 12:58
by Xu Liu, Tiantian Cong, Ping Liu and Peipei Sun

The Journal of Organic Chemistry
DOI: 10.1021/acs.joc.6b00097

25 Apr 12:53
by Keisuke Nogi, Tetsuaki Fujihara, Jun Terao and Yasushi Tsuji

Journal of the American Chemical Society
DOI: 10.1021/jacs.6b02961

19 Apr 12:34
by Keshav Raghuvanshi, Daniel Zell, Karsten Rauch and Lutz Ackermann

ACS Catalysis
DOI: 10.1021/acscatal.6b00711

19 Apr 12:33
by John J. Murphy
An important goal of modern organic chemistry is to develop new catalytic strategies for enantioselective carbon–carbon bond formation that can be used to generate quaternary stereogenic centres. Whereas considerable advances have been achieved by exploiting polar reactivity, radical transformations have been far less successful. This is despite the fact that open-shell intermediates are intrinsically primed for connecting structurally congested carbons, as their reactivity is only marginally affected by steric factors. Here we show how the combination of photoredox and asymmetric organic catalysis enables enantioselective radical conjugate additions to β,β-disubstituted cyclic enones to obtain quaternary carbon stereocentres with high fidelity. Critical to our success was the design of a chiral organic catalyst, containing a redox-active carbazole moiety, that drives the formation of iminium ions and the stereoselective trapping of photochemically generated carbon-centred radicals by means of an electron-relay mechanism. We demonstrate the generality of this organocatalytic radical-trapping strategy with two sets of open-shell intermediates, formed through unrelated light-triggered pathways from readily available substrates and photoredox catalysts—this method represents the application of iminium ion activation (a successful catalytic strategy for enantioselective polar chemistry) within the realm of radical reactivity.
Nature 532 218 doi: 10.1038/nature17438

12 Apr 08:42
by Mathias Paul, Martin Breugst, Jörg-Martin Neudörfl, Raghavan B. Sunoj and Albrecht Berkessel

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b13236

17 Mar 10:51
by Jones-Mensah, Ebenezer
Synthesis
DOI: 10.1055/s-0035-1560429

Dimethyl sulfoxide is generally characterized as a solvent and oxidant rather than as a substrate, building block, or synthon in organic chemistry. However, an abundance of reports have recently appeared that demonstrate dimethyl sulfoxide acting in these roles. This review article offers a comprehensive summary of the literature on this topic until the end of 2015. Synthetic transformations that have utilized the ‘C–S–C’, ‘C’, and ‘C–S’ fragments of dimethyl sulfoxide as building blocks are systematically summarized.1 Introduction2 History and Recent Highlights of DMSO-Based Oxidations3 DMSO-Based Methylthiomethylation (–CH2SMe)4 DMSO as a One-Carbon Synthon4.1 DMSO-Based Methylation (–Me)4.2 DMSO-Based Methylenation (–CH2–)4.3 DMSO-Based Annulation/Aromatization (=CH–)4.4 DMSO-Based Formylation (–CHO)4.5 DMSO-Based Cyanation (–CN)5 DMSO as Synthon for ‘S–C’ Functionalities5.1 DMSO-Based Thiomethylation (–SMe)5.2 DMSO-Based Methylsulfonylation (–SO2Me)6 Summary and Conclusions
[...]
© Georg Thieme Verlag Stuttgart · New York
Article in Thieme eJournals:
Table of contents | Abstract | Full text
17 Mar 10:35
New J. Chem., 2016, 40,2547-2553
DOI: 10.1039/C5NJ02314A, Paper
Dariush Khalili
Graphene oxide as a heterogeneous carbocatalyst catalyzes the direct thiocyanation of a variety of arenes and enolizable carbonyl compounds.
The content of this RSS Feed (c) The Royal Society of Chemistry

16 Mar 11:36
by Liyan Song, Xinqiang Fang, Zijia Wang, Kun Liu and Chaozhong Li

The Journal of Organic Chemistry
DOI: 10.1021/acs.joc.6b00008

16 Mar 11:32
by Yihua Sun, Hao Tang, Kejuan Chen, Lianrui Hu, Jiannian Yao, Sason Shaik and Hui Chen

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b12150