Journal of the American Chemical Society
DOI: 10.1021/jacs.5b05971
This review covers the formation of N- and S-containing heterocycles, initiated by gold-catalyzed nucleophilic attack of N- or S-nucleophiles onto alkynes. These types of nucleophiles have been somewhat overlooked as compared to their C- or O-counterparts in other reviews. In this particular work, their intramolecular gold-mediated attack onto alkynes is reviewed in depth. It is structured in such a fashion that the reader will get a clear view of which substrates react in which cyclization mode.




Fully stereodivergent dual-catalytic α-allylation of protected α-amino- and α-hydroxyacetaldehydes is achieved through iridium- and amine-catalyzed substitution of racemic allylic alcohols with chiral enamines generated in situ. The operationally simple method furnishes useful aldehyde building blocks in good yields, more than 99 % ee, and with d.r. values greater than 20:1 in some cases. Additionally, the γ,δ-unsaturated products can be further functionalized in a stereodivergent fashion with high selectivity and with preservation of stereochemical integrity at the Cα position.

Be selective: Ir-catalyzed allylic substitution of racemic allylic alcohols with chiral enamines generated in situ enables the fully stereodivergent α-allylation of protected α-amino- and α-hydroxyacetaldehydes. The method furnishes α-N/O-substituted γ,δ-unsaturated aldehydes in good yields and with excellent enantioselectivities. Phth=phthalimide; cod=1,5-cyclooctadiene; Ts=p-toluenesulfonyl; DCE=1,2-dichloroethane.


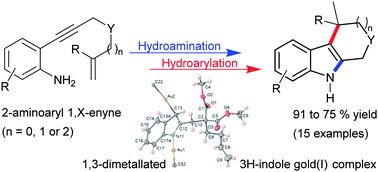
Removal of the chloride ligand from [AuCl(1-κP)] (2) containing a P-monodentate 1′-(diphenylphosphanyl)-1-cyanoferrocene ligand (1), by using silver(I) salts affords cationic complexes of the type [Au(1)]X, which exist either as cyclic dimers [Au(1)]2X2 (3 a, X=SbF6; 3 c, X=NTf2) or linear coordination polymers [Au(1)]nXn (3 a′, X=SbF6; 3 b′, X=ClO4), depending on anion X and the isolation procedure. As demonstrated for 3 a′, the polymers can be readily cleaved by the addition of donors, such as Cl−, tetrahydrothiophene (tht) or 1, giving rise to the parent compound 2, [Au(tht)(1-κP)][SbF6] (5 a) or [Au(1-κP)2][SbF6] (4 a), respectively, of which the last two compounds can also be prepared by stepwise replacement of tht in [Au(1-κP)2][SbF6]. The particular combination of a firmly coordinated (phosphane) and a dissociable (nitrile) donor moieties renders complexes 3/3′ attractive for catalysis because they can serve as shelf-stable precursors of coordinatively unsaturated AuI fragments, analogous to those that result from the widely used [Au(PR3)(RCN)]X catalysts. The catalytic properties of the Au-1 complexes were evaluated in model annulation reactions, such as the synthesis of 2,3-dimethylfuran from (Z)-3-methylpent-2-en-4-yn-1-ol and oxidative cyclisation of alkynes with nitriles to produce 2,5-disubstituted 1,3-oxazoles. Of the compounds tested (2, 3 a′, 3 b′, 3 a, 4 a and 5 a), the best results were consistently achieved with dimer 3 c, which has good solubility in organic solvents and only one firmly bound donor at the gold atom. This compound was advantageously used in the key steps of annuloline and rosefuran syntheses.

Self-stabilised catalysts: Dimeric and polymeric gold(I) complex cations with 1′-(diphenylphosphanyl)-1-cyanoferrocene ligand (L) coordinated as a hemilabile P,N donor, [Au(L-κ2P,N)]+, readily undergo cleavage of the Au![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) CN coordination bond and can be thus advantageously used as precursors for highly catalytically active, silver-free AuI species (see figure).
CN coordination bond and can be thus advantageously used as precursors for highly catalytically active, silver-free AuI species (see figure).
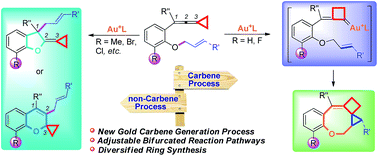
A detailed study of the gold-catalyzed tandem 1,3-carboxy migration/allene–enyne cycloisomerization was undertaken. It was found that after the initial allene formation the selectivity of the reaction is strongly influenced by the polarization of the remaining alkyne. Depending on the substitution pattern of the starting diynes, either a Schmittel- or a Myers–Saito-type cyclization was triggered. The 6-endo-dig Myers–Saito-type cyclization gave access to benzo[b]fluorenes, while the Schmittel pathway (5-exo-dig) delivered benzofulvenes as final products. In special cases a yet unknown pathway was opened by the ambiphilic nature of the allene moiety. In these cases completely different products were obtained by the nucleophilic attack of the alkyne moiety onto the allene that can also act as an electrophile. Mechanistic studies revealed that diradical pathways can be ruled out for this type of tandem cyclization reactions and it is shown that both steps of the reaction cascade are catalyzed by the gold complex.

Substitution patterns: A detailed study of the gold-catalyzed tandem 1,3-carboxy migration/allene–enyne cycloisomerization was undertaken. It was found that the selectivity of the reaction is strongly influenced by the polarization of the remaining alkyne. Depending on the substitution pattern of the starting diynes, either a Schmittel- or a Myers–Saito-type cyclization was triggered (see scheme).
Stereo- and chemodivergent enantioselective reaction pathways are observed upon treatment of alkylarylketenes and trichloroacetaldehyde (chloral) with N-heterocyclic carbenes, giving selectively either β-lactones (up to 88:12 dr, up to 94 % ee) or α-chloroesters (up to 94 % ee). Either 2-arylsubstitution or an α-branched iPr alkyl substituent within the ketene favours the chlorination pathway, allowing chloral to be used as an electrophilic chlorinating reagent in asymmetric catalysis.

Taming chloral two ways: Stereo- and chemodivergent enantioselective reaction pathways are observed upon treatment of alkylarylketenes and trichloroacetaldehyde (chloral) with N-heterocyclic carbenes (NHCs), giving selectively either β-lactones (up to 88:12 dr, up to 94 % ee) or α-chloroesters (up to 94 % ee). Either 2-arylsubstitution or an α-branched iPr alkyl substituent within the ketene favours the chlorination pathway (see scheme).
The reactions between terminal alkynes and α-chiral tosylhydrazones lead to the obtention of chiral pyrazoles with a stereogenic group directly attached at a nitrogen atom. The cascade reaction includes decomposition of the hydrazone into a diazocompound, 1,3-dipolar cycloaddition of the diazo compound with the alkyne, and [1,5] sigmatropic rearrangement with migration of the stereogenic group. This strategy has been successfully applied to the synthesis of structurally diverse chiral pyrazoles through α-chiral tosylhydrazones, obtained from α-phenylpropionic acid, α-amino acids, and 2-methoxycyclohexanone. Notably, the stereoretention of the [1,5] sigmatropic rearrangements represent very rare examples of this stereospecific transformation.

Maintaining a (con)figur(ation): Retention of configuration in [1,5] sigmatropic shifts are predicted by the Woodward–Hoffmann rules, but are not well-documented experimentally. Such shifts are observed in the reactions of α-chiral tosylhydrazones with terminal alkynes. The chiral pyrazole products are formed by a 1,3-dipolar cycloaddition and subsequent site-, regio-, and stereospecific [1s,5s] sigmatropic rearrangement. Ts=4-toluenesulfonyl.
A highly enantioselective gold(I)-catalyzed intermolecular annulation of 2-(1-alkynyl)-2-alken-1-ones with N-allenamides is presented. The present work represents the first example of a gold-catalyzed annulation with the proximal C![[DOUBLE BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8fe.gif) C bond of an N-allenamide, and is distinctly different from the previously observed annulations at the distal CC bond. Interestingly, both enantiomers of the products could be obtained in good yields with high regio-, diastereo-, and enantioselectivity by using either diastereomer of a binol-derived phosphoramidite as a chiral ligand.
C bond of an N-allenamide, and is distinctly different from the previously observed annulations at the distal CC bond. Interestingly, both enantiomers of the products could be obtained in good yields with high regio-, diastereo-, and enantioselectivity by using either diastereomer of a binol-derived phosphoramidite as a chiral ligand.

On the double: An unprecedented [3+2] annulation takes place with the proximal CC bond of N-allenamides in the presence of a chiral gold catalyst. Both enantiomers could be obtained in good yields with high regio-, diastereo-, and enantioselectivities by using either diastereomer of a binol-derived phosphoramidite ligand. binol=1,1′-bi-2-naphthol.
A gold(I)-catalyzed synthesis of indanones from trimethylsilylacetylenes and acylsilanes is presented. The reaction is initiated through a synergistic acylsilane activation–gold acetylide formation and involves consecutive alkyne σ-gold(I) addition, π-activation, and 1,2-migration of a silyl group. Studies performed on the reaction mechanism allowed to establish the nature of the silyl migrating group and invoke the participation of a gold(I) carbenoid intermediate. The reaction is completed by a gold(I) CH functionalization step.

The elegant way: A gold(I)-catalyzed synthesis of indanones from trimethylsilylacetylenes and acylsilanes was developed. The reaction involves a synergistic acylsilane activation–gold acetylide formation and consecutive alkyne σ-gold(I) addition, π-activation, and 1,2-silyl migration. Mechanistic studies suggest the participation of a gold(I) carbenoid intermediate.
The mechanisms of regiodivergent cyclizations of o-alkynylbenzaldehyde acetals and thioacetals catalyzed by Pd and Pt halides are studied. DFT calculations found that both reactions are initiated by electrophilic activation of the acetylenic moiety instead of the previously proposed metal-triggered CX (X=O, S) cleavage. Both the regioselective cyclization of the π-alkyne complex and the chemoselective [1,2]-migration in the carbenoid intermediate were determined as key steps to achieving the observed divergence. For acetal derivatives containing an internal alkyne, the 6-endo-dig cyclization is more favorable and leads to the carbenoid intermediate easily through further steps of CX fragmentation and carbocation cyclization. Then, from the carbenoid intermediate, the [1,2]-migration of sulfur is easier than that of H, Me, and Ph; whereas, a reversed aptitude was predicted for the oxygen analogue, which is consistent with the greater ability of sulfur atoms to stabilize β-carbocations. However, for precursors containing a terminal alkyne, the 5-exo-dig pathway is preferred and only the 1,2-disubstituted indene product is seen, irrespective of the nature of the acetal; thus, a different product from that reported in the literature is predicted for benzaldehyde acetal with a terminal alkyne at the ortho position. This prediction led us to reconsider some of the reported results and hidden realities were uncovered with solid new experimental evidence.

Where paths diverge: In-depth mechanistic understanding of the regiodivergent cyclizations of o-alkynylbenzaldehyde acetals and thioacetals catalyzed by Pd and Pt halides is achieved by combined theoretical and experimental studies (see scheme).
The reaction of [Au(C![[TRIPLE BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8fd.gif) Cn-Bu)]n with [Pd(η3-allyl)Cl(PPh3)] results in a ligand and alkynyl rearrangement, and leads to the heterometallic complex [Pd(η3-allyl){Au(CCn-Bu)2}]2 (3) with an unprecedented bridging bisalkynyl–gold ligand coordinated to palladium. This is a formal gold-to-gold transmetalation that occurs through reversible alkynyl transmetalations between gold and palladium.
Cn-Bu)]n with [Pd(η3-allyl)Cl(PPh3)] results in a ligand and alkynyl rearrangement, and leads to the heterometallic complex [Pd(η3-allyl){Au(CCn-Bu)2}]2 (3) with an unprecedented bridging bisalkynyl–gold ligand coordinated to palladium. This is a formal gold-to-gold transmetalation that occurs through reversible alkynyl transmetalations between gold and palladium.

Back and forth! Direct gold-to-palladium and reverse palladium-to-gold alkynyl transmetalations effectively produce a deceptively simple gold-to-gold transfer. The resulting bisalkynyl–gold complexes act as bridging ligands to palladium atoms in an unprecedented structural motive (see scheme).
 Open Access
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.



The gold-catalyzed 1,3-O-transposition reaction of ynesulfonamides provides a practical synthetic protocol for the synthesis of ynamides under mild conditions. This is the first 1,3-O-transposition example of ynesulfonamides in which a heteroatom is attached to the alkynyl terminal. A plausible mechanism is been proposed on the basis of 18O-labeling and control experiments as well as DFT calculation. By simple treatment, the obtained ynamides can easily be transformed into useful products.

The gold-catalyzed 1,3-O-transposition reaction of ynesulfonamides provides a practical synthetic protocol for the synthesis of ynamides under mild conditions. This is the first 1,3-O-transposition example of ynesulfonamides in which a heteroatom is attached to the alkynyl terminal.


Hydride abstraction from the gold (disilyl)ethylacetylide complex [(P)Au{η1-CCSi(Me)2CH2CH2SiMe2H}] (P=P(tBu)2o-biphenyl) with triphenylcarbenium tetrakis(pentafluorophenyl)borate at −20 °C formed the cationic gold (β,β-disilyl)vinylidene complex [(P)AuCCSi(Me)2CH2CH2Si(Me)2]+B(C6F5)4− with ≥90 % selectivity. 29Si NMR analysis of this complex pointed to delocalization of positive charge onto both the β-silyl groups and the (P)Au fragment. The C1 and C2 carbon atoms of the vinylidene complex underwent facile interconversion (ΔG≠=9.7 kcal mol−1), presumably via the gold π-disilacyclohexyne intermediate [(P)Au{η2-CCSi(Me)2CH2CH2Si(Me)2}]+B(C6F5)4−.

Good as gold: Cationic gold (β,β-disilyl)vinylidene complex 1 was generated by addition of a pendant silylium ion to the CC bond of a gold acetylide complex (see scheme, P=PtBu2(o-biphenyl)). The vinylidene C1 and C2 atoms of 1 undergo facile interconversion, presumably via a π-disilacyclohexyne intermediate. 29Si NMR analysis of 1 indicates delocalization of positive charge onto both the β-silyl groups and the (P)Au fragment.
Bonding and stabilizing effects in gold carbene complexes are investigated by using Kohn–Sham density functional theory (DFT) and the intrinsic bond orbital (IBO) approach. The π-stabilizing effects of organic substituents at the carbene carbon atom coordinated to the gold atom are evaluated for a series of recently isolated and characterized complexes, as well as intermediates of prototypical 1,6-enyne cyclization reactions. The results indicate that these effects are of particular importance for gold complexes especially because of the low π-backbonding contribution from the gold atom.

Is anybody there? Intrinsic bond orbital analyses based on DFT calculations on gold carbene complexes indicate little π backbonding from gold (A) and π stabilization from organic fragments even in cyclopropyl-substituted gold carbene complex intermediates (B, C).