Journal of the American Chemical Society
DOI: 10.1021/ja4111418

Heating a range of suitably substituted diazoacetoacetates produced a family of 2-(1,3-dioxolan-2-yl)phenyl ketenes that, under thermal conditions, smoothly underwent a [1,5]-H shift/6π-electrocyclic ring-closure sequence to give 1H-2-benzopyrans. The application of such processes to ketenes, produced by replacing the phenyl scaffolding with a thiophene ring, afforded thienopyrans. The aza-Wittig reaction of these 2-(1,3-dioxolan-2-yl)phenyl and thienyl ketenes with N-aryliminophosphoranes provided analogous ketenimines that transform into the respective 1(2H)isoquinolinones and thienopyridinones under similar thermal conditions, following the same type of cascade sequence.

Under mild thermal conditions, 2-(1,3-dioxolan-2-yl)phenyl ketenes and ketenimines convert into the respective spirocyclic 1H-2-benzopyrans and 1(2H)-isoquinolines by a [1,5]-H shift/6π-electrocyclization cascade sequence.
Belén Rubial:DDDD

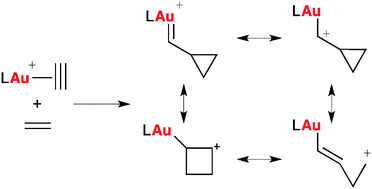
The front cover picture, provided by Belén Rubial, Alfredo Ballesteros and José M. González, graphically illustrates the ability of electrophilic gold to catalyze the addition of alkynylsilanes to aldehydes furnishing bis-alkynylation products, which has been demonstrated for the first time. The observed transformation is the result of two consecutive C![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) C bond-forming hits over a single carbon. This synthetic event results from a distinctive triggering device involving the switch from carbophilic to oxophilic activation control along the reaction path, as it is reported in the communication by Rubial, Ballesteros and González on pages 3337–3343 (DOI: 10.1002/adsc.201300578).
C bond-forming hits over a single carbon. This synthetic event results from a distinctive triggering device involving the switch from carbophilic to oxophilic activation control along the reaction path, as it is reported in the communication by Rubial, Ballesteros and González on pages 3337–3343 (DOI: 10.1002/adsc.201300578).

Belén RubialEsterificación con un fenol en la molécula.




Diene catalysts with a twist: The title C2-symmetric tetralin-fused 1,3-butadiene derivative is atropisomeric and can be resolved into the two helical enantiomers. The optically pure compound showed excellent enantioselectivity as well as unusually high catalytic activity as a chiral Lewis basic organocatalyst in the asymmetric allylation of various aldehydes with β-substituted allyltrichlorosilanes (see scheme).

Belén RubialCon amigüitos :)

A series of 2-(1,3-dioxolan-2-yl)phenylallenes that contained a range of substituents (alkyl, aryl, phosphinyl, alkoxycarbonyl, sulfonyl) at the cumulenic C3 position were prepared by using a diverse range of synthetic strategies and converted into their respective 1-(2-hydroxy)-ethoxy-2-substituted naphthalenes by smooth thermal activation in toluene solution. Electron-withdrawing groups at the C3 position accelerated these tandem processes, which consisted of 1) an initial hydride-like [1,5]-H shift of the acetalic H atom onto the central cumulene carbon atom; 2) a subsequent 6π-electrocyclic ring-closure of the resulting reactive ortho-xylylenes; and 3) a final aromatization step with concomitant ring-opening of the 1,3-dioxolane fragment. If the 1,3-dioxolane ring of the starting allenes was replaced by a dimethoxymethyl group, the reactions led to mixtures of two disubstituted naphthalenes, which were formed by the migration of either the acetalic H atom or the methoxy group, with the latter migration occurring to a lesser extent. Two of the final 1,2-disubstituted naphthalenes were converted into their corresponding naphtho-fused dioxaphosphepine or dioxepinone through an intramolecular transesterification reaction. A DFT computational study accounted for the beneficial influence of the 1,3-dioxolane fragment on the carbon atom from which the H-shift took place and also of the electron-withdrawing substituents on the allene terminus. Remarkably, in the processes that contained a sulfonyl substituent, the conrotatory 6π-electrocyclization step was of lower activation energy than the alternative disrotatory mode.

Keeping the heat on: Heating solutions of 2-(1,3-dioxolan-2-yl)phenylallenes at reflux in toluene caused their conversion into 1-(2-hydroxy)ethoxynaphthalenes through cascade processes that were initiated by a hydride-like [1,5]-H shift (see scheme, ERC=electrocyclic ring-closure). DFT calculations disclosed an interesting balance between the two rotatory modes of the 6π-electrocyclization step, depending on the substituents at the two linking carbon atoms.





Golden delivery: A gold(I)–NHC complex catalyses the protodecarboxylation of various (hetero)aromatic acids. This methodology is simple, highly efficient and combines both the advantages of copper and silver methods with applications to completely deactivated substrates. Moreover, intermediates in this reaction have been isolated and fully characterized and provide fundamental insights into the mechanism of protodecarboxylation with gold.

Beyond a doubt organocatalysis belongs to the most exciting and innovative chapters of organic chemistry today. Organocatalysis has emerged not only as a complement to metal-catalyzed reactions and to biocatalysis over the last decade, but also provides new asymmetric organocatalyzed reactions that cannot be accomplished by metal- or biocatalyzed reactions so far. A large number of organocatalytic processes are already well established in organic synthesis. Nevertheless, the number of publications in this field is still on the increase; new important results are produced constantly. This review gives a detailed overview of the latest developments and main streams in organocatalyzed asymmetric CC bond formation processes of the last three years. It is intended to outline the most important current findings focused on especially new synthetic methodologies.

A large number of organocatalytic processes are already well established in organic synthesis. Nevertheless, the number of publications in this field is still on the increase; new important results are produced constantly. This review gives a detailed overview of the latest developments and main streams in organocatalyzed asymmetric CC bond formation of the last three years. It is intended to outline the most important current findings focused on especially new synthetic methodologies.
Belén Rubial:D
The first successful gold(I)-catalyzed reaction of aryl aldehydes with trimethyl(arylethynyl)silanes to furnish bis-alkynylated derivatives is reported. Key CC bond-forming events involved in the catalytic cycle are analyzed.


A great leap forward toward the general synthesis of P-stereogenic compounds: Heating H3PO2 with (−)-menthol and paraformaldehyde gives easily crystallized menthyl hydroxymethyl-H-phosphinate (1). From this product, virtually any P-stereogenic compound can be synthesized (see picture).

Stronger acid, higher speed: The pKa values of a range of binol-derived Brønsted acids of three different types were measured and found to correlate directly with the catalytic properties of the acids: higher rate constants kI were observed for more acidic Brønsted acid catalysts (see plot; binol=1,1′-bi-2-naphthol).

No cycle required: The straightforward synthesis of acyclic (amino)(ylide)carbene gold complexes was achieved by reaction of isocyanide gold complexes with phosphorus and arsenic ylides as well as electron-rich olefins. Their ability to form bimetallic species and to act as ligand-transfer reagents has also been established.
A novel methodology based on a carbon-selective 1D HSQC experiment is proposed for the determination of 1H–19F coupling constants. The method offers rapid access to 1H–19F coupling constants that cannot be directly measured from the 1H NMR spectrum due to the complexity and/or broadening of the proton signals or because the values are very small. As an additional benefit, the method also provides the relative sign of the 1H–19F coupling constant.

A variant of the 1D HSQC experiment in which the offset of the selective carbon pulse is set to the frequency of a peak of a fluorinated carbon atom showing the α or β spin state of the fluorine provides the magnitude and sign of 1H–19F coupling constants.