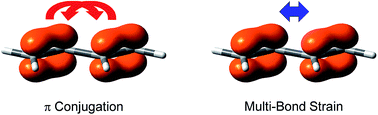Journal of the American Chemical Society
DOI: 10.1021/jacs.5c00974

 Open Access
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
Nature, Published online: 15 April 2025; doi:10.1038/d41586-025-01124-w
Some studies have received hundreds of thousands of citations, Nature’s updated analysis shows.Nature, Published online: 15 April 2025; doi:10.1038/d41586-025-01126-8
An analysis for Nature reveals the studies that appear most in the reference lists of current publications.Nature, Published online: 15 April 2025; doi:10.1038/d41586-025-01125-9
A Nature analysis reveals the 25 highest-cited papers published this century and explores why they are breaking records.Nature, Published online: 15 April 2025; doi:10.1038/d41586-025-01204-x
Ensuring that conferences are inclusive requires diverse organizers
Open Access   This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.

A nickel-catalyzed group-exchange strategy has been developed for the direct conversion of aromatic lactones into cyclic hemiboronic acid bioisosteres. Scope evaluation and product derivatization experiments demonstrate broad functional-group compatibility and the synthetic value of this strategy. Furthermore, the application of this methodology to the rapid modification of lactone cores in bioactive molecules underscores its practical utility.
Bioisosteric replacement is an important strategy in drug discovery and is commonly practiced in medicinal chemistry; however, the incorporation of bioisosteres typically requires laborious multistep de novo synthesis. The direct conversion of a functional group into its corresponding bioisostere is of particular significance in evaluating structure-property relationships. Herein, we report a functional-group-exchange strategy that enables the direct conversion of aromatic lactones, a prevalent motif in bioactive molecules, into their corresponding cyclic hemiboronic acid bioisosteres. Scope evaluation and product derivatization experiments demonstrate the synthetic value and broad functional-group compatibility of this strategy, while the application of this methodology to the rapid remodeling of chromenone cores in bioactive molecules highlights its utility.
Open Access  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
Open Access  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.


Reactions of the dimeric alkaline earth hydrides, [(BDI)AeH]2 (BDI=HC{(Me)CNDipp}2; Dipp=2,6-i-Pr2C6H3; Ae=Mg, Ca), with Ph3GeH in the presence of THF provide H2 and [(BDI)AeGePh3(THF)]. Although the synthesis of the magnesium derivative is complicated by THF ring opening, the calcium reagent is a viable source of the triphenylgermanide nucleophile.
The dimeric calcium and magnesium hydrides, [(BDI)AeH]2 [BDI=HC{(Me)CNDipp}2, Dipp=2,6-i-Pr2C6H3; Ae=Mg or Ca] do not react with Ph3GeH in non-coordinating solvent. Addition of THF, however, induces deprotonation and access to monomeric Ae-germanide complexes, [(BDI)Ae{GePh3}(THF)], both of which have been structurally characterized. Although this process is facile when Ae=Ca, the analogous magnesium-based reaction requires heating to temperatures >100 °C, under which conditions germanide formation is complicated by THF ring opening and the generation of an alkaline earth germyl-C-terminated n-butoxide, [(BDI)Mg{μ2-O(CH2)4GePh3}]. Reactions of [(BDI)Ca{GePh3}(THF)] with N,N′-di-isopropylcarbodiimide and benzophenone provide the respective germylamidinate and germylalkoxide derivatives, [(BDI)Ca{(i-PrN)2CGePh3}(THF)] and [(BDI)Ca{OC(GePh3)Ph2}(THF)], demonstrating its potential as a well-defined and soluble source of the [Ph3Ge]− anion in nucleophilic addition reactions.

Reactions of [(2,6-Dipp2C6H3)Sn(μ-H)]2 (Dipp=2,6-i-Pr2C6H3) with [(BDI)AeH]2 (BDI=HC{(Me)CNDipp}2; Ae=Mg, Ca) provide heterobimetallic bis-μ2-hydrido species, [[(BDI)Ae-μ-(H)2-Sn(C6H3-2,6-Dipp2)]. Although both compounds react with hex-1-ene to provide alkaline earth derivatives of tetravalent organostannides, the magnesium reagent promotes twofold addition of the alkene whereas the calcium-based reaction is arrested after a single hydrostannylation event.
Reactions of a m-terphenylhydridostannylene with β-diketiminato magnesium and calcium hydrides provide bis-μ2-hydrido species, the heterobimetallic constitutions of which are maintained after the addition of THF donor solvent. In both cases, reactions with hex-1-ene result in the formation of tetravalent organostannyl alkaline earth derivatives. Whereas the magnesium reagent undergoes facile twofold addition, the calcium-centered process is arrested after a single alkene reduction event. This contrasting behavior is assessed to result from the heavier alkaline earth element's ability to form a persistent polyhapto interaction with an arene substituent of the m-terphenyl ligand.




Michael Cowleynice

Alkali metals in water are always at the brink of explosion. Herein, we show that this vigorous reaction can be kept in a non-exploding regime, revealing a fascinating richness of hitherto unexplored chemical processes. A combination of high-speed camera imaging and visible/near-infrared/infrared spectroscopy allowed us to catch and characterize the system at each stage of the reaction. After gently placing a drop of a sodium/potassium alloy on water under an inert atmosphere, the production of solvated electrons became so strong that their characteristic blue color could be observed with the naked eye. The exoergic reaction leading to the formation of hydrogen and hydroxide eventually heated the alkali metal drop such that it became glowing red, and part of the metal evaporated. As a result of the reaction, a perfectly transparent drop consisting of molten hydroxide was temporarily stabilized on water through the Leidenfrost effect, bursting spectacularly after it had cooled sufficiently.

The vigorous reaction of alkali metals in water was kept in a non-exploding regime through gently placing a drop of Na/K alloy on water. Initially, solvated electrons are produced (blue color), before the exoergic reaction leading to H2 and OH− heats the drop such that it becomes red, and part of the metal evaporates. A transparent drop of molten hydroxide is then temporarily stabilized on the water through the Leidenfrost effect before bursting spectacularly.

Open Access  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.

As a result of the increased polarity of the metal–carbon bond when going down the group of the periodic table, the heavier alkali metal organyl compounds are generally more reactive and less stable than their lithium congeners. We now report a reverse trend for alkali metal carbenoids. Simple substitution of lithium by the heavier metals (Na, K) results in a significant stabilization of these usually highly reactive compounds. This allows their isolation and handling at room temperature and the first structure elucidation of sodium and potassium carbenoids. The control of stability was used to control reactivity and selectivity. Hence, the Na and K carbenoids act as selective carbene-transfer reagents, whereas the more labile lithium systems give rise to product mixtures. Additional fine tuning of the M−C interaction by means of crown ether addition further allows for control of the stability and reactivity.

Heavy but stable: The reactivity of s-block metal organyl compounds generally increases when going down the group of the periodic table, often limiting applications of the heavier congeners. However, the reverse of this trend has now been demonstrated for alkali metal carbenoids by replacement of Li by Na or K. The resulting compounds exhibit an increased thermal stability, which allows their isolation and selective application.
Michael Cowleygood pone to share Steph. Look at the compound they propose in this paper... they are bonkers! First one to propose a synthetic route to one of them wins... something (the chance to make it?)