Hi everyone: I’ve written a long deep-dive on the present state of the McMansion, from farmhouse chic to imminent environmental collapse. If you’ve been seeing an inordinate number of big ugly houses pop up in your neighborhood, you are not alone!
Over the last week, three U.S. banks have failed. More banks are under extreme stress. This stress is not new and was not unknown but is becoming common knowledge rapidly. We may be in the early stages of a banking crisis.
This situation is evolving very rapidly and this essay will not. Please check the WSJ or Financial Times for updates on the fluid bits. Hopefully this essay helps contextualize what is reported.
“Crisis” is a bit of a strong word, even when invoked as a potential outcome, and I try to be fairly sober-minded. I’d like to explain how we got here, how the relevant institutions are generally expected to work, what seems to be different this time, and what smart people who are not normally professionally engaged in this might find relevant to know about the infrastructure that we all depend on.
Short disclaimer: I worked at Stripe (which is not a bank, but works with many banks) for six years prior to leaving full-time employment recently. I am an advisor there now. My views are entirely my own, and my analysis is only informed by publicly available data. I put a longer disclaimer at the bottom.
Why are banks failing?
As we previously covered in a discussion about deposit insurance, now unfortunately topical, banks do not fail in a day. The seeds of their destruction are sown and watered for years, and then they are reaped quickly.
Importantly, these do not say “seeds of destruction: definitely don’t plant these!” on the package. People have a great desire for there to be a narrative here; for a bank failure to require stupidity or malfeasance or ideally stupid malfeasance.
The thing killing banks is a very simple idea with profound consequences. It is not a secret: when interest rates rise, all asset prices must fall. This is both almost a law of nature and also perpetually underestimated in how much it affects the world outside of asset prices. For example, in January 2020, I pointed out that obviously engineering compensation includes an interest rate derivative, because it includes equity. This is very not obvious to many people in tech, including financially sophisticated people!
But equity is not the only thing that embeds an interest rate derivative. All prices embed an interest rate derivative. The price of eggs embeds an interest rate derivative, among many other things, like how it reflects the cost of grain. The price of grain embeds an interest rate derivative. The world sits atop four elephants who stand astride the risk-free rate, and then it is interest rates all the way down.
The price of eggs, and other important parts of the consumer basket, is a major contributing reason why we are here. The United States (through the Federal Reserve) made a considered decision to manage inflation by hiking interest rates. That is, explicitly, an intervention to push down the price of eggs (and other things), via a lever which happens to be much more amenable to direct action than other available levers for controlling egg prices. This lever can be applied across the entire consumer basket in parallel. And so it was.
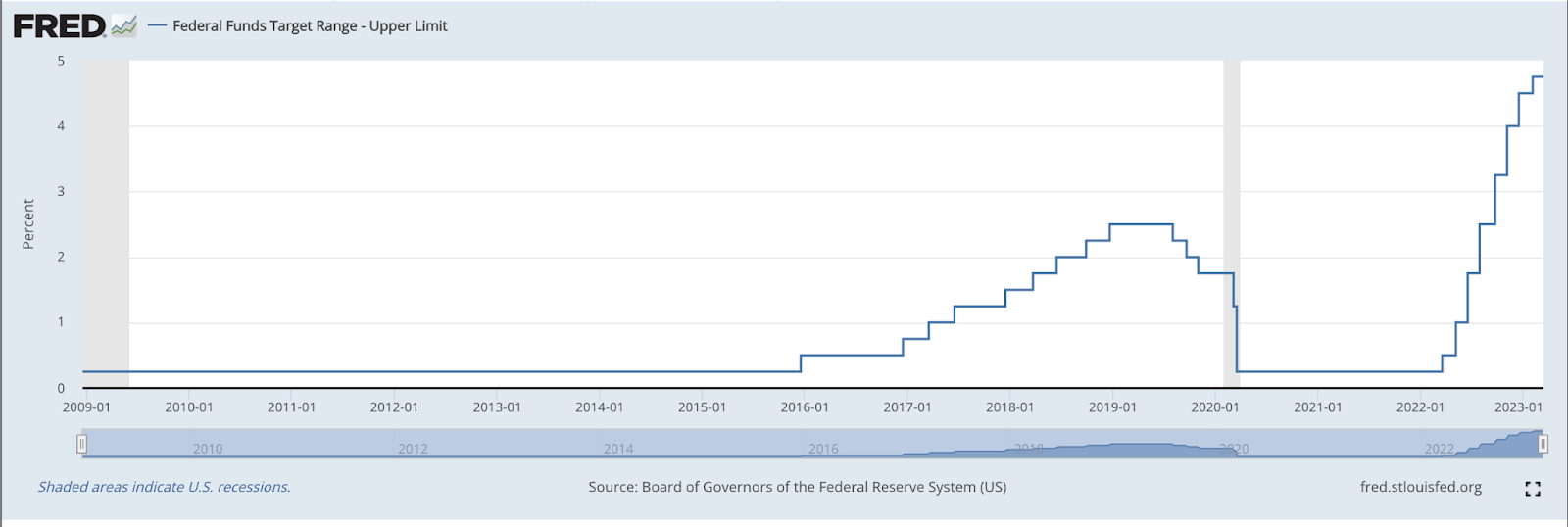
If you recall the ancient history of *checks notes* the past 15 months, we went from a regime where prevailing interest rates were just above zero to almost 5%. This was the most aggressive hike in rates since World War II, or to put it another way, in the history of the modern economic order.
Federal Funds Target Range, per the Federal Reserve
The decision to sharply manage down the price of eggs was, indirectly but inescapably, also a considered decision to cause large notional losses to all holders of financial assets. That includes everyone with a mortgage, every startup employee with equity, and every bank.
That is the proximate cause of the banking crisis, if in fact we are in a crisis. Three banks failed first, because for idiosyncratic reasons they were exposed to sudden demands for liquidity, which makes large declines in the value of one’s assets unsurvivable. But there are many more banks which have a similar issue on their balance sheet.
A useful heuristic from bond math
I apologize for a very 101-level financial math lesson but it’s unavoidable, useful, and may not have featured in your education (and, of course, Matt Levine beat me to mentioning it): there is a heuristic for the value of bonds.
Every bond and instrument created on top of bonds has a “duration”, which you can round to “how much time left in years until we expect this to be paid back?” And every bond and instrument on top of bonds has its market price move down by 1% per year of duration if interest rates move up 1%, and vice versa. (There is better math available but this is math you can trivially perform in your head, and is close enough to blow up large portions of a financial system.)
So if you held ten year bonds and interest rates went up 4% in a year, your ten year bonds are down, hmm, somewhere in the 35%ish range. This is true regardless of whether the bonds are good bonds. If you want to sell them today, the people buying them have better options than you had a year ago, and to induce them away from those better options you have to give them a 35%ish discount.
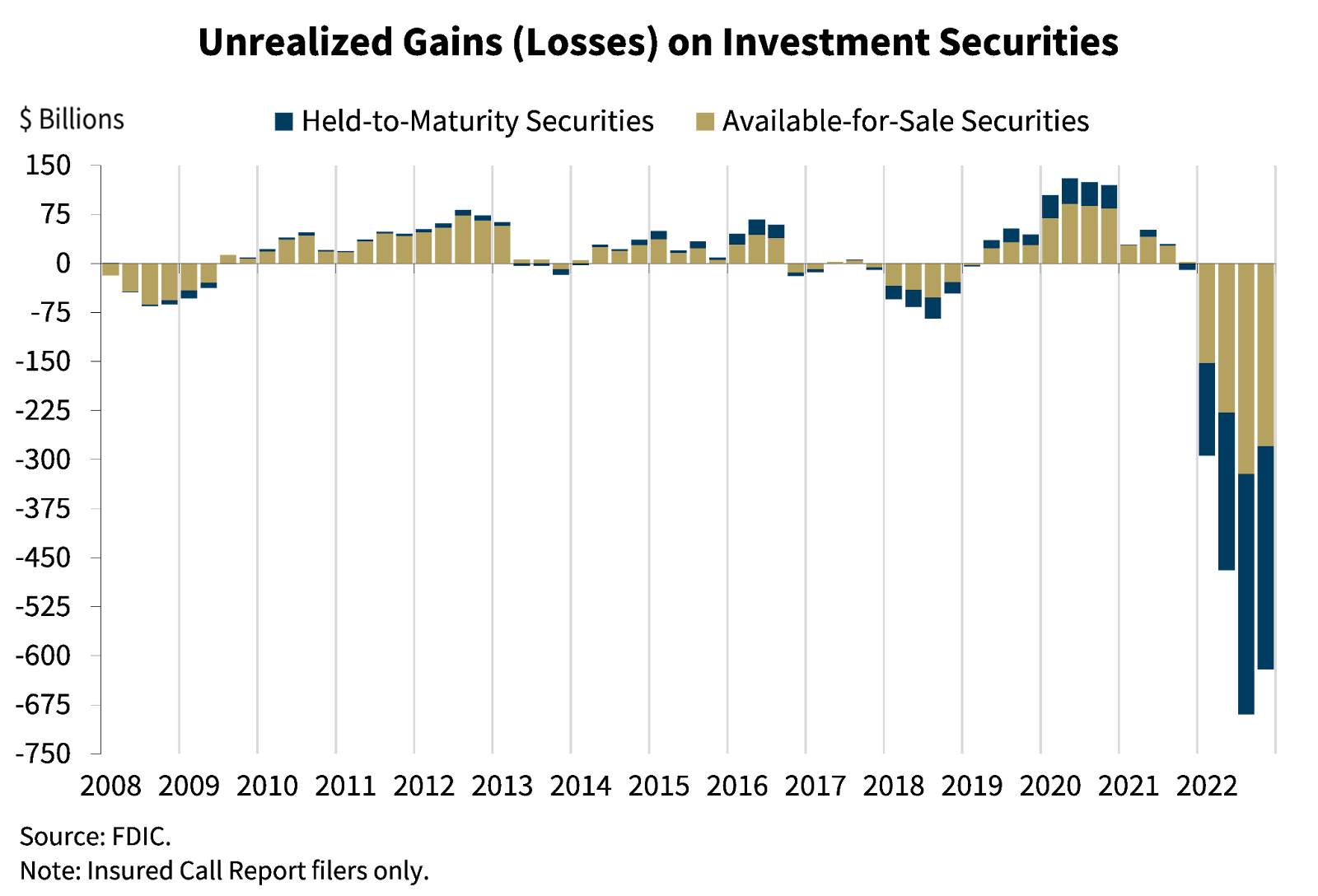
We now come to one of the most important charts in the financial world, courtesy of the FDIC in February:
The U.S. banking system has $620 billion in unrealized losses on investment securities, per the FDIC
$620 billion. The U.S. banking system lost $620 billion. Six hundred twenty billion dollars. That is a loss no less real than if money had been loaned out to borrowers who defaulted. It might be temporary! If interest rates go down, bond prices will recover. (And sometimes defaulting borrowers receive an inheritance or get bailed out! But one doesn’t generally want to count on that.)
Was this because the banks invested in poor credit? No. The price of everything embeds an interest rate derivative, including definitionally perfect credit like U.S. Treasuries. The type of security most numerically relevant here is functionally immune to credit risk: agency-issued mortgage-backed securities.
You might remember that financial instrument from 2008. Many people are going to fixate on that coincidence far more than is warranted. In 2008 those embedded bad-and-mispriced credit risk which had an uncertain backstop. In 2023 the losses are caused by a bad-and-mispriced interest rate risk with a rapidly evolving backstop. But all asset prices include an interest rate derivative.
“Why do banks buy exotic assets with lots of letters in the name, like MBS from GSE? Why can’t they just do banking? Like, make regular loans to real people and businesses with income to service them? That would surely solve this, right?”
It would not. If they created loans with fixed rates, just plain vanilla loans warehoused on their own balance sheet in the “traditional business of banking”, the rate environment would have exactly the same effect. It already has had this effect.
In addition to the $620 billion in losses in securities, there exist staggering losses in the loan books of every bank that wrote fixed rate loans in 2021. And 2020. And 2019. And 2018. And 2017. And 2016. And 2015. And 2014. And 2013. And 2012. And 2011. And 2010. And 2009.
Most people sensibly don’t care about any of this, and only care when a financial product which is core to their lives—bank deposits—suddenly and unexpectedly ceases to function. Bank deposits are much more complicated products than they are believed to be. When banks fail, the most important societal impact is that deposits, which are money no less real than physical script and in many ways much more real, suddenly have an unanticipated risk of not being money.
Maturity transformation
What is the connection between deposits, bank runs, and the value of ten year bonds in conditions of rising interest rates? I’m glad you asked.
You pay an explicit bill to most businesses which provide you valuable services. You get deposits for free*, emphasis on the asterisk. The tellers and the lawyers and the engineers and the regulators and the insurance company and the equity providers who collectively must labor diligently to give you deposits still need to get paid. They get paid largely by harvesting the option value from depositors as a class and creating something new out of it.
Banks engage in maturity transformation, in “borrowing short and lending long.” Deposits are short-term liabilities of the bank; while time-locked deposits exist, broadly users can ask for them back on demand. Most assets of a bank, the loans or securities portfolio, have a much longer duration.
Society depends on this mismatch existing. It must exist somewhere. The alternative is a much poorer and riskier world, which includes dystopian instruments that are so obviously bad you’d have to invent names for them.
Take an exploding mortgage, the only way to finance homes in a dystopian alternate universe. It’s like the mortgages you are familiar with, except it is callable on demand by the bank. If you get the call and can’t repay the mortgage by the close of the day, you lose your house. What did you do wrong to make the mortgage explode? Literally nothing; exploding mortgages just explode sometimes. Keeps you on your toes.
Exploding mortgages don’t exist and can’t exist in our universe. But it is important that, from a bank’s perspective, the dominant way people bank sometimes explodes. That asymmetry is the mismatch. We expect banks to manage this risk, and we expect society to tolerate it (and sometimes cover the bill for it), because exploding mortgages are worse than this risk.
We have moved some of this mismatch out of the banking system, by e.g. securitizing mortgages and selling them to pension funds which can match them against natural liabilities (e.g. actuarial tables of when pensioners will retire and require their payouts). But the banking system holds a lot of duration mismatch risk, and likely always will.
This is, like all the other risks to banks, something which is managed and regulated. Sometimes management screws up or priorities their bonuses over prudential risk mitigation. Sometimes regulators are, feel free to choose your phrasing, asleep at the switch or not sufficiently empowered.
Can I excerpt that FDIC speech from three weeks ago? While the FDIC obviously must moderate their public comments, this is the payload:
> Unrealized losses on available–for–sale and held–to–maturity securities totaled $620 billion in the fourth quarter, down $69.5 billion from the prior quarter, due in part to lower mortgage rates. The combination of a high level of longer–term asset maturities and a moderate decline in total deposits underscores the risk that these unrealized losses could become actual losses should banks need to sell securities to meet liquidity needs.
The sacred duty of equity is to protect depositors from losses. After it is zeroed, the losses must come from somewhere. We do not celebrate equity getting vaporized, except insofar that sacrifice of oneself in satisfaction of a duty to others is generally praiseworthy, but we certainly want to be aware that it happened.
The world is, belatedly, realizing that this did actually happen. Past tense.
This realization creeped in around the edges with e.g. Byrne Hobart on February 23rd noting that one of the U.S.’s largest banks was recently technically insolvent but almost certainly in a survivable way. And, to be fair, a few short funds and the Financial Times had come to this realization a bit before Byrne. Then, a few weeks later, the entire financial system almost simultaneously discovered how much they doubted precisely one half of his thesis.
I submit to you that the regulators probably did not understand a few weeks ago that this situation was factually as concerning as it is.
Don’t read this as a statement about competence or the lack of it; just read it as a factual claim about the constitution of the Problem Bank List. The Problem Bank List is figurative state secret, specifically to prevent inclusion on the PBL from causing a run on the bank if it were to become common knowledge.
At least one bank which failed last week was not a Problem Bank three weeks ago. Reader, that should not ever happen.
“How do you know this if the Problem Bank List is a state secret?” Because they report the aggregate total of the assets of all banks on the list and publicly available data plus math a 4th grader can do in their head suffices to prove this claim.
Finance is an industry with many smart people in it. The same goes for regulatory agencies. You’re welcome to your guess of how many of them asked a 4th grader “Were all the banks which failed this week on the Problem Bank List or do we have an unknown unknown?” prior to reading this paragraph.
There exists this same problem at banks that are not on the Problem Bank List. I would normally hedge that sentence with something like “likely”, but the market has woken up and is now aggressively repricing risk and publishing findings. Those findings are deeply concerning and, for social reasons, I must direct you to the financial media of your choice to read them.
We went multiple years without a bank failure, of any size, in the United States. We then had three in a week, including one (by some measures) larger than any during the last financial crisis. It would take a very brave and confident person to forecast no additional bank failures in the next two weeks. It would take a very interestingly calibrated person to say that, contingent on there being a bank failure, that that bank must necessarily have been on the Problem Bank List.
Liquidity problems are the proximate cause of bank failures
The reason for relative sanguinity about unrealized losses in the banking sector denominated in the hundreds of billions to low single digit trillions of dollars, and forgive me for harping on that fact but it is a fact about the world we live in, is that banks do not need to pay out all deposits simultaneously. Functionally no bank anywhere could do that, and the theoretical exception is considered not desirable as a matter of public policy and therefore does not exist.
Banks designate certain assets on their books as “available for sale”, those which they expect to perhaps sell to raise liquidity, and “held to maturity.” Losses in the ATS portfolio are relatively noisy, because they immediately ripple into one’s income statement, are reported quarterly, and are extremely salient for all stakeholders. Losses in the HTM securities are basically fine until they aren’t.
This isn’t entirely because management prefers to keep its head in the sand. Banks are institutions designed to exist over timelines longer than interest rate cycles. This implies certain assets of theirs will always be underwater and certain assets of theirs will always be “worth more than we paid for them.” To the extent that the bank is simply holding the asset to collect the income from it this all comes out in the wash. The day-to-day movements are in normal times a distraction and get relegated to a footnote.
We do not expect the footnote to swallow the bank, and that is an important update to our model of the world. We do not expect it to swallow multiple banks. We do not expect to not have a high-quality estimate for how many banks it will swallow in the next two weeks.
The three bank runs which already happened had idiosyncratic causes, but “if accounted for accurately, the bank is insolvent” is the sort of thing which, if one stipulates to it, one would suggest might generate bank runs in the near future. And so there was a policy response, which much commentary has assumed is primarily about the banks which no longer exist, and the satisfaction of their depositors, and which is actually much more about banks in danger which might yet be saved.
Trying to forestall a banking crisis
The losses banks have taken on their assets are real. They already happened. They are survivable if banks remain liquid.
The Federal Reserve, Department of the Treasury, and Federal Deposit Insurance Corporation released a joint statement over the weekend to adjust people’s expectations regarding banks that still exist. The key element of the response is a temporary extension of credit to banks collateralized by high-quality assets at their par value, rather than their market value. This is called the Bank Term Funding Program.
The hope is that a bank facing liquidity pressure could tap this credit program, in addition to existing credit programs and source of liquidity, and thereby avoid a downward spiral of selling assets, realizing losses, pushing asset prices down, spooking markets and depositors, and repeating at a very high cycle rate until the bank doesn’t exist.
We recently went through that cycle faster than we thought possible with regards to a bank which responsible people considered very safe. According to the official record, one of the institutions went from being financially healthy one day to insolvent the next. I believe that narrative to be face-saving, but it is what The System currently is messaging as the truth, so let’s accept it for now. If this is the truth, what unfortunate truths might we learn in the near future?
This is a temporary program; banks can only tap this liquidity for about a year. In the ordinary course, bank runs don’t last for a year; they either cause an institution to fail very quickly or peter out. But the other reason this is time-bounded is to defang the moral hazard, on behalf of both banks and their customers. (Moral hazard in insurance is when the existence of insurance makes it incentive-compatible for you to be imprudent in your own risk taking, expecting someone else to bear the consequences.)
Banking regulators want banks to take the strong medicine solution to the problem.
If banks have experienced hundreds of billions to single digit trillions of dollars in losses, realized or no, they have a very limited set of options. Hoping for a miracle is one. Experiencing a sudden dramatic shift downwards in interest rates, which would cause them windfall gains for exactly the reason they experienced windfall losses, is another. Grinding out many years of profits in the ordinary business of banking to fill the hole is a third.
But the thing which is actually within their immediate ability and control is simple and painful. The sacred duty of equity is to take losses before depositors do. Equity has taken losses. Depositors must be shielded. Equity must be raised to take the losses again.
Equity, of course, has a choice in a free market system as to which risks it wants to take. It flowed into banks in good times at prices banks were reasonably happy with. They now need to raise in what is no longer a good time, at prices banks (and existing equity holders, etc) will not be happy with, because the new marginal equity appreciates the risk environment it is entering more than the equity raised a while ago.
This is the short explanation for why bank stocks are getting hammered right now. A share is a one-over-some-denominator claim on the equity of the bank. Sophisticated people are realizing that the numerator is lower than they expect and the denominator is shortly to be larger, and potentially much larger, than they expect. Existing shares are perforce worth less than they were before we woke up to this realization. Banks will need to go to the market to sell new shares at these less favorable prices.
Count this as another knock against the strong-form efficient market hypothesis. None of these dynamics are particularly complicated by the standards of finance. The core facts are not secrets; they were exhaustively disclosed on a quarterly basis. Charts were made.
Anyone could have made a killing if they put two and two together even a week ago. A killing was, mostly, not made. (Killings perhaps remain available as of this writing, if that is your thing.)
Deposit insurance expansion
Bank deposits in the U.S. are insured up to $250,000 per depositor per account type per institution. The exact definition of “account type” is a sort of wonky detail; just assume it is $250,000 historically per depositor/institution pair and you’ll save some braincells for the meatier issues.
By special and extraordinary action, the FDIC has announced that two recent bank failures will backstop all deposits, not just all insured deposits. Much commentary has focused on the decision to create winners out of losers vis depositors at those two institutions.
This is an effect of the policy but is neither the intent nor the rationale.
Let me speculate about some things which may have happened this weekend, with arbitrarily high confidence.
Over the weekend, the regulators made some calls and asked regional banks what deposit outflows looked like on Friday and how many wires were queued up for execution Monday morning. This was complicated by some banks finding it surprisingly difficult to add numbers quickly. You see, the core puts the queued wired requests in a different part of the system than Friday’s outflows. We have a report of Friday outflows, but it gets crunched by an ETL job which only finishes halfway through Saturday, and Cindy who understands all of this is on vacation, and… and eventually very serious people said Figure Addition The #*(%#( Out And Call Me Back Soonest.
Regulators then heard the numbers, did a bit of modeling in Excel, and then went into wartime execution mode. Regulators have, of course, not declared this war, because it is a war on the public’s perception of reality, and to declare war is to surrender.
The $620 billion in losses on securities and the concomitant loss on loans is not distributed evenly across the U.S. banking sector, but it is distributed across the U.S. banking sector. Every institution thanking its risk managers for them having a below-average amount of it implies that some other institution has more of it.
And so we are in a situation where some institutions, whose names are not yet in headlines but may be very shortly indeed, are under acute stress. And we are also beginning to understand a mechanism by which a handful of institutions fell off a precipice, where we understand the edge of that precipice to be eroding, because we currently believe interest rates will go up again. (That belief is shifting rapidly; the rapid decline in 2 year Treasury yields is a sign that the markets are adjusting expectations and beginning to doubt the forecast future sharp hikes.)
Financial institutions are also adjusting to the new reality rapidly. Over the weekend, like every other customer of a particular bank, I got an email from the CEO explaining that they had ample liquidity but had just secured a few tens of billion of additional liquidity, prudent risk management, no problems here, all services are as up as ever, yadda yadda yadda.
Securing more liquidity may be prudent, and the announcement of securing liquidity may be prudent, but this is not an email you send to all customers in good times. Banks typically take communications advice from the Lannisters: anyone who needs to say they have adequate liquidity does not have adequate liquidity. History is replete with examples. Bank CEOs know this. They know their sophisticated customers know this. And yet that email was still written, reviewed by management and crisis comms and counsel, and then sent.
Deposit insurance also some legacy issues
Deposit insurance is an important piece of social technology, and so successful that some believe that it is the primary reason deposits are safe. It is, of course, the backstop to the primary things which make deposits safe, which is the ordinary risk management of banks, a complex and mostly effective regulatory regime, and $2.2 trillion of private capital that signed up to be incinerated if there are faults in earlier controls. The deposit insurance fund, by comparison, is about $130 billion, which you can compare to that $620 billion in losses number prior to thanking capital for its service to society.
But, much like we’ve previously talked about how credit cards are legacy infrastructure, deposit insurance is also legacy infrastructure. It is designed to adjust the expectations of large numbers of relatively slow-acting low-sophistication users by credibly dampening the pain to “regular users of the banking system” that banking stress threatens.
But the world deposit insurance now protects is different than the one it was developed in, and I think it may need to be updated. One much remarked upon elsewhere is that some banks have hypernetworked customer bases who can through relatively independent action tweet and WhatsApp themselves to withdrawing $42 billion in a day.
But deposit insurance is institutionally aware that some institutions have concentrated deposits and lots of deposits are controlled by sophisticated actors. We had capital-intensive businesses with chainsmoking professionals who'd prefer their businesses to survive a bank run during all the relevant crises. The architects of deposit insurance knew these people exist and that they were a primary vector for runs historically. This problem is planned for. It was not created by Twitter.
Let's talk about the problem it doesn't institutionally prepare for. The entire edifice of deposit insurance rests on the assumption the primary harm from bank failure, at least that worthy of societal attention, falls first on direct depositors of the bank and secondly on spillover stress in the rest of the system.
This is a reasonable model, and like all models it is wrong but useful.
Consider the case of Rippling, a startup I have no affiliation with. Rippling has a complicated business; one portion of that is being a payroll provider. Payroll providers, as a type of business, are much older than iPhones but effectively younger than many policy measures designed to mitigate banking crises. (Rippling is a tiny one; some exist in the Fortune 500.)
When Rippling’s bank recently went under, there was substantial risk that paychecks would not arrive at the employees of Rippling’s customers. Rippling wrote a press release whose title mostly contains the content: “Rippling calls on FDIC to release payments due to hundreds of thousands of everyday Americans.”
Prior to the FDIC et al’s decision to entirely back the depositors of the failed bank, the amount of coverage that the deposit insurance scheme provided depositors was $250,000 and the amount it afforded someone receiving a paycheck drawn on the dead bank was zero dollars and zero cents.
This is not a palatable result for society. Not politically, not as a matter of policy, not as a matter of ethics.
Every regulator sees the world through a lens that was painstakingly crafted over decades. The FDIC institutionally looks at this fact pattern and sees this as a single depositor over the insured deposit limit. It does not see 300,000 bounced paychecks.
Payroll providers are the tip of the iceberg for novel innovations in financial services over the last few decades. There exist many other things which society depends on which map very poorly to “insured account” abstraction. This likely magnifies the likely aggregate impact of bank failures, and makes some of our institutional intuitions about their blast radius wrong in important ways.
What would happen if my bank were to go into receivership this weekend?
We covered this previously, but the dominant answer historically is that it is sold and you have a new bank on Monday with functionally nothing else changing. The system has worked very well; we have gone years since the last bank failure, most failures are small, most are entirely resolved by the following Monday, and even deposits over the limits held at banks which failed have rarely taken losses over the last few decades. On the few occasions they have, those losses have been miniscule.
The system recently looked at the combination of published rules, availability of a transaction over the weekend, degree of surprise, and preparedness of suitors… and it blinked, because of what it could actually have delivered on Monday (yesterday).
That would have been full satisfaction of insured deposits, perhaps fifty cents on the dollar satisfaction of uninsured deposits, and a few months of uncertainty as to the timing and level of eventual satisfaction for the remainder. Actual losses would have probably been zero or a few cents on the dollar, eventually, probably.
That resolution is a much worse resolution than the one the system typically obtains and it would have affected many more people than is typical. This may be, if not the new normal, a new concerning potential recurring pattern during uncertain times.
People may have a mental model that a bank keeps a list of all its customers and can therefore quickly calculate e.g. who is insured and to what degree, so that it can pass this list to the FDIC, so that those people can get their money on Monday. This is a useful mental model for first approximations and does not actually describe the world you live in.
For example, FDIC insurance insures the “actual owners” of accounts, and not the entities those accounts are titled to. One important type of account which exists in the world is the For Benefit Of (FBO), where someone might hold money in trust for someone else in their own name.
FBO aren’t newfangled things dreamt up in Silicon Valley. Trusts as an institution date back to the middle ages; regulations have successfully anticipated how they used to be used.
Decades ago, the dominant mental image people might have had for FBO accounts was Lawyer Larry holding a settlement on behalf of Client Carla because lawyers are more like banks than regular people are like banks. The FDIC insures Carla, not Larry, even if Larry has fifty Carlas commingled in a single account and the bank only knows them as “names available on request.” (This is perhaps surprising for people who think banks need to Know Your Customers. The bank customarily adheres to its written policy about KYC for FBOs. Their regulator is OK with the policy. All of this is the normal business of banking and entirely uncontroversial.) To make Carla whole, it has to learn Carla exists first, which implies a process that cannot conclude by Next Monday.
Well that’s an edge case, right. Lawyers and FBO accounts have to be a teeny tiny percentage of all deposits and, while this would be greatly inconvenient for Carla, presumably if she is still banking through her lawyer in 2023 she is rich and sophisticated.
Let’s talk about fintech.
Many fintech products have an account structure which looks something like this sketch: a financial technology company has one or several banking relationships. It has many customers, enterprises which use it for e.g. payment services or custodying money. Those services are not formally bank accounts, but they perform a lot of feels-quite-bankish-if-you-squint to the people who rely on them to feed their families. The actual banking services are provided to those users by the banks, who are disclosed prominently on the bottom of the page and in the Terms and Conditions.
Each enterprise has their own book of users, who might number in the hundreds of thousands or millions, in a single FBO account at the bank, titled in the name of the enterprise or the name of the fintech. The true owners of the funds are known to the bank to be available in the ledgers of the fintech but the bank may have sharply limited understanding of them in real-time.
And so I ask you a rhetorical question: is this structure robust against the failure of a bank handled other-than-cleanly, such that, come the following Monday, those users receive the insurance protection which they are afforded by law? Mechanically, can that actually be done? Is our society prepared to figure that out over a weekend? Because during this past weekend, that sketch I wrote out about banks being confuddled by addition for a few hours almost certainly happened.
There are a sharply finite number of hours between Friday and Monday and we cannot conveniently extend them to cover multiparty discussions about how to get a core system to import a CSV dumped by a beleaguered data scientist from Jupyter based on a hopefully up-to-date MongoDB snapshot so that it can be provided to the FDIC agents on site.
I am very frustrated by political arguments about desert, which start with an enemies list and celebrate when the enemies suffer misfortune for their sins like using the banking system.
Be that as it may: most enemies lists do not include taxi drivers, florists, teachers, plumbers, etc etc you get the drift literally every strata of society is exposed to products which bank for them in complicated ways. These people will be hurt by bank failures. We as a society do not accept this, which is a large portion of why they are protected if they bank directly with a financial institution, and why we promise they are protected if their money is in a more complicated account structure.
I am very sure our society and institutions are operationally capable of delivering on the promised and counted-upon protection for some of the ways these depositors access banking services.
Many people who read this might feel a bit of negative surprise that structures like this sketch exist in the world and are deployed pervasively. ("Was that allowed?! Where were the regulators?" Yes. The usual places.)
Interestingly, that has not been the dominant worry about the adequacy of deposit insurance in the fintech industry. The dominant worry, among clueful people on this narrow and wonky topic, has been that deposit insurance would not protect some people exposed to structures where the bank survived but the fintech did not.
Given this worry, fintechs trumpeting FDIC insurance to mean that users faced de minimis risk of loss of funds felt like misselling what they were offering.
The good news: it seems like the problem we’re immediately faced with is the sort of thing that deposit insurance actually insures against: the failure of financial institutions. The hypothetical losses would be covered. The bad news: banks are failing and more may fail, potentially including some banks with customers that have business models younger than Its A Wonderful Life (1946).
It is not obvious to me that people, including people in positions of authority and responsibility, understand that society has wandered its way into commitments shaped like this one. But it has, and so maybe they should (while dealing with the other fires) seek to gain more understanding of the current operation of financial infrastructure that is pervasively deployed and pervasively relied upon by many people, including arbitrarily sympathetic people.
Not that I think someone needs to be sympathetic to be worth a duty of care here. Infrastructure undergirds society; failures of it are a per se emergency. Anyone who cheers an infrastructure failure because of the first order consequences of it will find themselves negatively surprised.
What should users of the banking system do?
I suggest that you go to someone who actually has a professional duty of care to you, but that feels unsatisfying, and so let me make some general observations.
One is that the banking system is more resilient than appreciated, even under conditions of immense stress. From the perspective of a typical consumer using the banking system, you can probably blithely ignore that this is happening. Nightmares for systemic stability might be utterly non-events for you personally.
To the extent one wants to take low-cost actions one is unlikely to regret, I would suggest one has at least one backup financial institution. If one hypothetically does not, I would observe that opening bank accounts rounds to free. Thousands of perfectly good financial institutions exist. If one were to put money into a backup account, perhaps enough money to get through a weekend or to get through a payroll cycle, one would have access to money even if one’s primary financial institution was unexpectedly unavailable for a short time due to serious issues. (Having credit available at diverse institutions is, of course, another option.) This has the added benefit of helping if the issue is, for example, total computer failure at the bank rather than financial catastrophe. It has been known to happen.
If one has more money in a financial institution than applicable insurance limits, and one does not have a professional advisor about that money, and one does not feel capable of confidently answering questions about their risk management, one should probably find a clueful advisor. I have no particular advice on sorting clueful advisors from many who passed the relevant exams, charge outrageously, and know even less about this subject than non-experts currently Googling while stressed.
My observations for businesses would be more complicated.
Many people believe that businesses should have a treasury department who considers liquidity and risk management to be literally the only thing they do. That sounds great in theory, but in the world we actually live in, you will actually hire a treasury department afew hundred employees after your bank account is above FDIC coverage limits. (Deposit insurance was designed for a world with sharply different employment patterns!)
And so, if you are a founder in the substantial chunk of the economy between those two goalposts, you should breathe a sigh of relief that the FDIC and other regulators are going into crisis management mode.
Many banks and technology firms have, and some will quickly rush to market, various automated treasury management solutions. These do some of the work of a treasury department, at a tiny fraction of the cost of expensive professionals.
It seems to be popular right now to shame businesses and suggest they need to manage the counterparty risk their bank represents. This is actually advocacy for the most sophisticated and largest financial firms in the world to have a new high-margin revenue stream renting this solution to the substantial fraction of the economy too large to benefit from deposit insurance and too small to hire a treasury department.
The basic offering here, which I will avoid endorsing any particular provider of, is “We will establish relationships with N financial institutions in parallel. We automate money movement between them on your behalf, such that you can treat your money as being in one logical pile. However, at legally relevant times, in legally relevant ways, you only have a maximum of $250,000 in each institution. This will allow you to effectively 5X or 10X or… well there are thousands of banks and we are tireless in finding partners… the deposit insurance limit. This will cost you money, just like all financial services cost you money, and it may or may not be 100% obvious exactly how much money it costs you.”
I will note that there is an interesting policy angle on whether we, as a society, would prefer for deposit insurance to be effectively unlimited if and only if one is smart enough to pay a software company (or financial services firm, but I repeat myself) to do this for you.
In addition to “treasury management”, sometimes firms phrase this offering as “cash sweep”, which I mention in case you’re wondering what words you need to say to a salesman to get the pitch. The offering largely does what is says on the tin. Despite the above policy response, I’d expect the salesmen of it to be booked beyond capacity signing up new customers this week, at every firm that has it in-market and at some which are rushing to fix their lack of it.
Any parting thoughts?
The banking system is well-regulated, resilient, and strong. Most institutions in the U.S. are comfortably OK at the moment. Some may well not be. Failures, and particular surprising failures, in heavily interconnected core infrastructure have a worrisome tendency to cascade.
This is not the end of the world, but the last five days (!) include a material and negative update on our understanding of the state of the world. It has surprised many people at many different institutional vantage points who would expect to not be surprised by this exact issue.
You’re probably going to end up hearing a lot more about this. If for some reason you don’t read Byrne Hobart or Matt Levine, fix that. For breaking news, your financial news outlet of choice will be all over this for the foreseeable future. I recommend moderating one’s degree of reliance on group chats or Twitter, less because they are likely to be less accurate than media coverage (very not obvious to me) and more because your degree of risk here is likely lower than justifies 24/7 monitoring of this situation unless you have reasons why that is obviously not the case.
A long and boring disclaimer relegated to a footnote
Market observers have a purity ritual where they exhaustively disclaim whether they have financial interests in stocks they are discussing. I think that’s irrational in my case but rituals are useful things, so here’s a longer disclosure statement than you probably want:
I don’t and won’t short bank stocks, mostly because it’s impossible to do when keeping my nose provably clean given my position in the information graph. I do invest in individual bank stocks, but not materially (they’re a sixth of the economy and maybe 1% of the part of my portfolio I can conveniently price?), and for an idiosyncratic reason.
My life is weird by the standards of retail bank consumers—”business owner with American citizenship plus Japanese residence” puts me in a reference class of only a few hundred people banked in either nation. Banks will routinely steamroll reference classes of a few hundred people, by accident. You can buy a bank’s attention to bespoke needs by bringing it deposits, but it takes a lot more money than I have. Or you can buy a trivial number of shares of the bank and call Investor Relations if you have any problems.
This is one of many fun hacks I picked up over the years as an unpaid advocate for people with routine banking issues. Customer Service might fob off a retiree who wants a NSF fee reversed. Investor Relations, on the other hand, is socialized to guess that anyone calling it is more likely to be a pension fund manager and less likely to be a pension fund beneficiary. And so they can use very free calendars, no managed-to-the-minute-CS-drone quota, and substantial organizational heft to escalate things to any department on your behalf, with the implicit endorsement that Capitalism Called And It Requires You Resolve This Immediately.
Another weird thing about me: some people collect baseball cards. I collect bank accounts. I never set out to do this but by the time I realized I had a collection I had some borderline rational reasons for not reversing the decision, like “well I have to understand professionally how banking apps work and it is useful to have a survey of them installed” and “if I ever lose a U.S. bank account as a non-resident opening a new one is a pain in the keister so maybe I should have, oh, five backups... per account type... including for my LLC... in each country."
And so, when you combine these two facts, I am directly exposed to a lot of bank stocks, but in relatively tiny amounts. This includes banks under substantial stress. I have not sold and have not changed my banking as a result of risk of failure.
Why do I believe this is an irrational disclosure, despite general support for this ritual? Because I live in a society, which is sufficient information for you to know that I’m structurally levered long to the stability of the banking system, much like you are.
by Travis Okulski on Road & Track, shared by Patrick George to Jalopnik
Mr. Regular was asked to be a celebrity judge at Corvettes at Carlisle. Before he went, he wrenched his back, meaning he had to do it under the influence of steroids and painkillers. Here’s his drug-addled recollection of what happened.
Meet Jumpy the dog. This dog can jump higher than you, skateboard better than you, dive better than you, walk on its front paws better than you, surf better than you, catch a Frisbee better than you, do a backflip better than you, and ride a scooter better than you. Jumpy is better than you.
Ok, this dog is a better skateboarder, but Jumpy is still better than him. Jumpy is better than everyone. (thx, dad)
In a surprise twist, a creationist accuses scientists of argument from authority and trying to hide contradictions with inscrutable jargon. Transubstantial!
I've mentioned this before but it bears repeating. One of my Ph.D. students (Sharon Shtang) wrote her thesis on sequence comparisons and phylogenetic trees. She found a quotation from Emil Zuckerkandl and Linus Pauling in their 1965 review. They were commenting on using amino acid sequences to prove evolution. This seemed at the time to be an example of overkill since evolution was then, and is now, a fact. They said ...
Some beating of dead horses may be ethical, where here and there they display unexpected twitches that look like life.
Evolutionists reluctantly admit most evolution is free of selection and therefore non-Darwinian (neutral evolution). When pressed, they’ll say neutral drift is real, but they don’t like it when the dots are connected in a way that demonstrates neutral evolution refutes Darwinism, that there is a contradiction between Dawkins’ vision and neutral evolution! The way Darwinists deal with this violation of the law of non-contradiction is to pretend no contradiction exists. They’ll obfuscate and fog the issue with myriad technical terms and irrelevancies so that the illusion of non-contradiction is protected from public view. Confusion and the illusion of some higher knowledge are their friends, clarity and education of the public are their enemies.
If Dawkins had been faithful to the facts, he wouldn’t have even written The Blind Watchmaker because population genetics precludes his vision of evolution from being reality in anything but his silly Weasel simulations.
There is a simple formula from Wiki that says the rate of new mutations is the rate at which new mutations become features of every member of the population (a process called fixation).
The population size is N and the Greek symbol μ (mu) is the mutation rate.
It stands to reason a slightly deleterious mutation is almost neutral, hence, approximately speaking the rate that slightly deleterious mutations become part of every member of the population is on the same order of the slightly deleterious mutation rate. That means if every human is getting 100 dysfunctional mutation per generation, about 100 dysfunctional mutations are getting irreversibly infused into humans every generation (a ratchet so to speak).
But as bad as that is, it’s actually worse in reality. Remember broken bacterial parts in anti-biotic resistance, or blindness in cave fish, or sickle cell anemia? Those are “beneficial” (in the Darwinian sense) mutations, but destructive in the functional sense. So it is actually generous the creationists are modeling the dysfunctional mutations as slightly deleterious (whereas a fair argument might actually model some of the dysfunctional mutations as “beneficial”). So the creationists are cutting Darwinists a lot of slack, and yet, even then the dysfunctional mutations will get fixed (become members of all individuals) in a population! Not to mention, lots of bad may get purged from a population only to get replaced with new generations of bad....
But obvious math is something Darwinism hates dealing with! The above equation should be painful evidence against evolution being some process of increasing complexity from a primordial virus to incredible minds like Newton or Einstein. Darwinist won’t come to terms with it, they won’t come to terms with even a computer simulation based on population genetic models. Oh well! But anyway, Christopher Rupe and John Sanford will be presenting the results of a computer simulation that illustrates the above equation. It’s sort of like beating a dead horse or beating living puppies. It’s not very sporting, but Darwinists keep propping up that dead horse for creationists to keep beating.
Zuckerkandl, E. and Pauling, L. (1965) in EVOLVING GENES AND PROTEINS, V. Bryson and H.J. Vogel eds. Academic Press, New York NY USA
Thirty, almost forty, years ago when zebrafish were an up and coming model system and very few labs were working on them, we were used to going to conferences and reciting the zebrafish litany, a list of attributes that justified us working on such an oddball animal: we’d explain, for instance, that it was prolific, fast developing, and optically transparent, so we could see right into the nervous system in the living embryo. And you know, not once have I ever been asked the really simple and obvious question: if it’s transparent, then how do we see anything inside it?
I know. It takes a moment to step outside the assumptions (we say we see stuff, after all) and think about what transparency actually means. Of course, I always have an answer prepared: “With differential interference contrast microscopy, Nomarski optics specifically.” A polysyllabic phrase is always an effective way to shut down the rubes, isn’t it?
But I can also explain it in simple English, especially if I can flail about on a chalkboard at the same time. So let’s try.
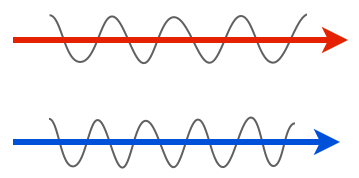
We’ll start with the very minimal basics. Light is a wave, and the properties of that wave are what our eyes see. For instance, one property of a wave is amplitude: in the two light rays below, the lower one has a larger amplitude, so we’d see it as having a greater intensity, or being brighter, than the top one.
Another property is wavelength or frequency. Shorter wavelengths (higher frequencies) have have more energy than longer wavelengths (lower frequencies), and we see these differences as color. In the cartoon below, the longer wavelength of light would be seen as red, while the shorter one is blue.
Those are the characteristics of light that our eyes can detect, so when I say that zebrafish embryos are transparent, I mean that light passing through them does not exhibit a significant reduction in amplitude or a change in wavelength — it does not get significantly dimmer nor does it change color.
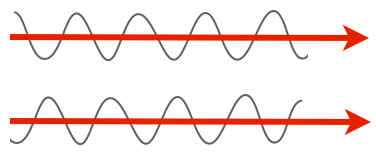
But there are other important properties of light. One is phase. Notice how in the previous picture of the amplitude, the peaks in one line up with the peaks in the other, and the troughs line up with the troughs? We’d say those two rays of light are in phase. They don’t have to be, though. For instance, here are two rays of light that are completely out of phase, with the peaks of one lining up with the troughs of the other.
Here’s the thing, though: our eyes can’t discriminate the phase of light. Both of those beams have the same amplitude and the same wavelength, so they’d look completely identical to us. Which is a shame, because lots of transparent things shift light phases, and especially of interest, lots of biological materials shift the phase. All you need to do is have a material with a different refractive index and you’ll get a phase shift.
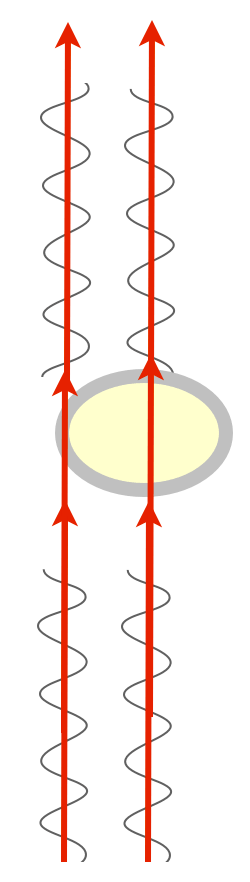
For example, most of a cell is water, and it’s surrounded by a membrane that is mostly oil/fats. These have different refractive indices. Shine two beams of light on a transparent cell, one that passes through the center (mostly water) and one that passes tangentially through the membrane (lots of oil), and you end up with two beams of light that now have different phases. Which we can’t see, darn it. My zebrafish embryos are full of membranes in complex arrangements, so while light passes through them with amplitude and wavelength (intensity and color) relatively unaffected, the phases of light are all scrambled and cockeyed in interesting ways full of information. We just have to have a way to detect phase and we could see lots of phenomena in the embryo.
There is a way to visualize the relative phase of two beams of light, though. If you could just bring them together, combine them, they could interfere with one another. If the two beams are in phase, peaks lining up with peaks, they’d sum up and you’d see a greater amplitude — the combined beam would be brighter. This is called constructive interference. If they’re out of phase, peaks lining up with troughs, they’d cancel each other out, they’d destructively interfere, and you’d get a dimmer beam.
We have this really clever way of combining two beams of light: they’re called prisms and lenses, which bend light. All we have to do is shine parallel beams of light through our specimen, and then use prisms to bend one and bring it into alignment with the other, and we’d get interference patterns that change the intensity of the light, and we’d be able to detect phase changes!
There is a tricky bit to this, though. This is a way of comparing the phase of two rays of light; what are we going to compare? One technique is to illuminate with ring of light, and use light that hasn’t passed through your specimen as a reference beam, and compare that to light perturbed by your specimen, the specimen beam. This is the technique used in standard phase contrast microscopy. It has some drawbacks I won’t get into in any detail; you get halos that reduce the resolution of your image. We don’t like halos. It’s kind of like the lens flares that cheesy sci-fi cinematographers sprinkle into their movies.
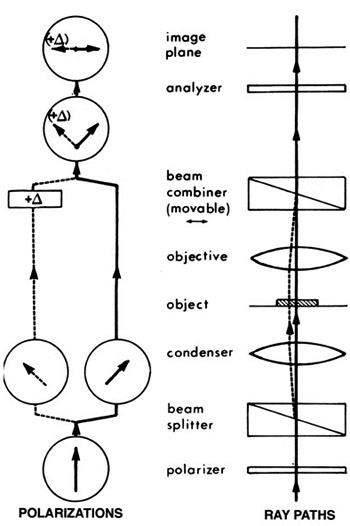
Differential interference contrast uses a different method to generate a reference beam. We illuminate with two beams of light that have different polarity and that are offset slightly (by a fraction of a micrometer) from one another. Polarity is another of those properties of light that our eyes can’t really detect; Dr Dryskull has an excellent post on polarity, but simply put, those waves of light aren’t all necessarily vibrating in the same plane — they can be wiggling in all sorts of directions. We use a Wollaston prism, a piece of crystal that splits light by its polarity, so that we have light vibrating in one plane, and another beam vibrating at 90° to the first, both shining through the specimen in parallel. Their phase will be affected in different ways because each will be passing through a very slightly different part of the specimen. Then they are recombined by passing through a second Wollaston prism, allowing them to interfere with one another, to produce interference patterns.
Does this diagram help? Basically you have all kinds of glass in the condenser and above the lens that jigger the heck out of light to split, isolate, and recombine the beams into visible patterns of constructive and destructive interference, allowing you to visualize shifts in phase, which represent changes in the refractive index, of cells and tissues.
More simply put, this technique compares the phase shift of each point on the specimen to a point 0.2 microns to the right (or whatever; we have knobs that let us shift the relative orientation). If you’re looking at a uniform field, this comparison yields the same intensity everywhere; but at regions in transition, for instance where phase is changing rapidly at a membrane boundary, you get bright and dark edge enhancement. This turns a bland, flat field of cells into a picture of clustered basketballs with a kind of lovely shadowing effect. Like so:
It can also go horribly awry, but I did this to make the difference even more obvious. Here is a field of embryonic cells in a zebrafish imaged without phase optics — we’re just seeing the subtle shifts in intensity of the light as it passes through. (If you’re interested, you’re seeing the margin of the expanding epiblast in a pre-gastrula embryo, with the yolk syncytial layer at the bottom of the field.)
Now what I’ve done is slipped in the various prisms and polarizers for DIC, and jacked the contrast up to 11. It’s ugly, but you should be able to see the vivid difference you can get by playing with phase; in particular, notice the great big YSL nuclei at the bottom right. The point here is that we can visualize all kinds of transparent objects in the embryo just by adjusting the widgets on the scope.
There are drawbacks. For one, all that glass diminishes the light intensity at every step — you need a good bright light source to start with. It takes some finesse to use properly (see photo above), and increases the complexity of configuring the scope by quite a bit. I’ve tried to train many students to use this stuff, and most of them throw up their hands in horror and get completely confused — too many knobs! Too finicky! It requires esthetic judgment! Good grief, it’s like…art or something.
This is the usual state I find my microscope in. Those of you who know what I’m talking about will see instantly that both the Wollaston prism and analyzer sliders have been pulled out of the light path. I usually feel like I’ve done well if I teach them to adjust the condenser properly. Grumpy aside: have you noticed how often student scopes have no adjustable condenser at all? You don’t know how to use a microscope if you don’t know how to set up Köhler illumination.
One last picture to show you that DIC can look very pretty. This is the tail bud of a 21 hour embryo; you can see somites with myoblasts, the notochord, mesenchyme, all kinds of cool stuff if you know what you’re looking at. And you can see it all in a transparent embryo.
One of the joys of preserving my own food is having delicious, locally grown summer fruits year-round. But as wonderful as traditional fruit preserving methods are, I can only eat so much jam. So I also pickle some of my blueberries, mulberries, cherries, strawberries, peaches, and other summer fruits. Pickling is an incredibly easy, safe, and, it turns out, delicious way to preserve fruit. Plus, in addition to the tasty orbs of fruit, you also create flavorful liquids that are perfect for adding to homemade cocktails or sodas. Everything in the jar has a purpose.
This blueberry pickle recipe uses traditional warming spices and red wine vinegar, which give the fruit and liquid a rich, earthy flavor. The pickled berries and liquid can be used on ice cream, in pancakes and muffins, or with a cheese board. They can also be used to make blue sunrise cocktails, a twist on the classic mimosa. Enjoy as a refreshing drink right now or in the dead of winter as a tasty reminder of summer’s bounty.
Sweet Blueberry Pickle
Yield: About 2 pints Note: You will need two one-pint jars and some cheesecloth.
2 cups blueberries
1½ cups red wine vinegar
1 cup sugar
¼ teaspoon salt
¼ teaspoon vanilla
1 cinnamon stick
½ teaspoon whole cloves
½ teaspoon whole allspice
Tie cinnamon stick, cloves, and allspice into a piece of cheesecloth.
In a medium saucepan, bring vinegar, sugar, salt, vanilla, and spice bag to a boil, stirring to dissolve sugar. Reduce heat, cover the pan, and simmer for 10 minutes.
Meanwhile, pack one cup of blueberries into each pint jar.
Remove spice bag from pickling liquid and discard. Ladle hot pickling liquid into jars to cover blueberries. (They will float, and that’s okay.)
If canning, process jarred blueberries in a hot water bath for 10 minutes. Otherwise, cover jars with a loose lid and allow to cool before refrigerating. Pickled fruit will keep in the refrigerator for about year.
Blueberries and their liquid will taste best if you can allow them to sit for a day or two before eating.
Blue Sunrise Cocktail
1 oz. blueberry pickle liquid
½ oz. elderflower liqueur
2 oz. orange juice
6 oz. dry champagne
Pickled blueberries and candied orange peel for garnish
In a Collins glass, stir blueberry pickle liquid, elderflower liqueur, and orange juice together with ice. Top with champagne. Garnish with pickled blueberries and candied orange peel.
Seems to have failed on my "kept-unread" stream, which is possibly the most important thing missing from the Takeout archive. But it still seems like it's worth running.

 Are your eyes good enough to see the Crab Nebula expand?
Are your eyes good enough to see the Crab Nebula expand? 








 I've mentioned this before but it bears repeating. One of my Ph.D. students (Sharon Shtang) wrote her thesis on sequence comparisons and phylogenetic trees. She found a quotation from Emil Zuckerkandl and Linus Pauling in their 1965 review. They were commenting on using amino acid sequences to prove evolution. This seemed at the time to be an example of overkill since evolution was then, and is now, a fact. They said ...
I've mentioned this before but it bears repeating. One of my Ph.D. students (Sharon Shtang) wrote her thesis on sequence comparisons and phylogenetic trees. She found a quotation from Emil Zuckerkandl and Linus Pauling in their 1965 review. They were commenting on using amino acid sequences to prove evolution. This seemed at the time to be an example of overkill since evolution was then, and is now, a fact. They said ...