Shared posts
[Research Article] Heads-up limit hold’em poker is solved
Gold(I)-Catalyzed Intermolecular Cycloaddition of Allenamides with α,β-Unsaturated Hydrazones: Efficient Access to Highly Substituted Cyclobutanes

Metal-Free Enantioselective Electrophilic Activation of Allenamides: Stereoselective Dearomatization of Indoles
Abstract
The effective and unprecedented chiral BINOL phosphoric acid catalyzed (1–10 mol %) dearomatization of indoles through electrophilic activation of allenamides (ee up to 94 %), is documented. Besides the synthesis of 3,3-disubstituted indolenine cores, a dearomatization/hydrogen transfer cascade sequence is also presented as a new synthetic shortcut toward highly enantiomerically enriched indolines.

Breaking the aromaticity: The enantioselective dearomatization of indoles with allenamides is achieved through Brønsted acid catalysis. A range of 3,3-disubstituted indolines as well as indolenines is synthesized in a highly chemo-, regio-, and stereoselective manner.
Mechanistic Studies on the Rearrangement of 1-Alkenyl-2-alkynylcyclopropanes: From Allylic Gold(I) Cations to Stable Carbocations
Abstract
An allylic gold(I) cation, proposed as key intermediate in the gold-promoted rearrangement of 1,5-enynes bearing a fixed conformation, has been detected and characterized by NMR spectroscopy. Moreover, its participation in the overall transformation was confirmed. Computational studies indicate that the gold-catalyzed transformation occurs through an uncommon rearrangement. Additionally, this study led us to isolate and characterize a stable homoantiaromatic carbocation.

No longer elusive: The allylic gold(I) cation 2, which has been proposed as an intermediate in the rearrangement of alkynylcyclopropanes (1) into alkynylcyclohexadienes (3), has been detected and characterized by NMR spectroscopy. Participation of 2 was supported experimentally and theoretically, and through these studies, a stable homo-antiaromatic carbocation was isolated and characterized.
Explanation of Counterion Effects in Gold(I)-Catalyzed Hydroalkoxylation of Alkynes

Gold-Catalyzed Intermolecular Synthesis of Alkylidenecyclopropanes through Catalytic Allene Activation
Abstract
A stereoselective gold(I)-catalyzed intermolecular cyclopropanation of allenamides with stabilized sulfonium ylides is reported. This transformation enables the direct synthesis of diacceptor alkylidenecyclopropanes and proceeds under very mild conditions through allene activation.

Three is my lucky number: A mild stereoselective gold(I)-catalyzed intermolecular cyclopropanation of allenamides with stabilized sulfonium ylides is reported. This transformation delivers diacceptor alkylidenecyclopropanes and unusually proceeds through allene activation rather than by metallocarbene formation (see scheme, EWG=electron-withdrawing group).
Synthesis, Structure, and Reactivity of a Gold Carbenoid Complex That Lacks Heteroatom Stabilization
Abstract
Hydride abstraction from the neutral gold cycloheptatrienyl complex [(P)Au(η1-C7H7)] (P=P(tBu)2(o-biphenyl)) with triphenylcarbenium tetrafluoroborate at −80 °C led to the isolation of the cationic gold cycloheptatrienylidene complex [(P)Au(η1-C7H6)]+ BF4− in 52 % yield, which was characterized in solution and by single-crystal X-ray diffraction. This cycloheptatrienylidene complex represents the first example of a gold carbenoid complex that lacks conjugated heteroatom stabilization of the electron-deficient C1 carbon atom. The cycloheptatrienylidene ligand of this complex is reactive; it can be reduced by mild hydride donors, and converted to tropone in the presence of pyridine N-oxide.

Hydride abstraction from the neutral gold cycloheptatrienyl complex 1 (P=P(tBu)2 (o-biphenyl)) with Ph3C+ BF4− formed the cationic gold cycloheptatrienylidene complex 2, which was characterized by single-crystal X-ray diffraction. The cycloheptatrienylidene ligand of 2 is reactive; it can be reduced by mild hydride donors, and converted into tropone in the presence of pyridine N-oxide.
[Report] Regioselective ketone α-alkylation with simple olefins via dual activation
Nucleophilic Addition/Double Cyclization Cascade Processes between Enynyl Fischer Carbene Complexes and Alkynyl Malonates

Isolation of a Non-Heteroatom-Stabilized Gold–Carbene Complex
Abstract
Gold–carbene complexes are essential intermediates in many gold-catalyzed organic-synthetic transformations. While gold–carbene complexes with direct, vinylogous, or phenylogous heteroatom substitution have been synthesized and characterized, the observation in the condensed phase of electronically non-stabilized gold–carbenes has so far remained elusive. The sterically extremely shielded, emerald-green complex [IPr**Au=CMes2]+[NTf2]− has now been synthesized, isolated, and fully characterized. Its absorption maximum at 642 nm, in contrast to 528 nm of the red-purple carbocation [Mes2CH]+, clearly demonstrates that gold is more than just a “soft proton”.

Gold: more than just a “soft proton”: The preparation of the gold complex [IPr**Au![[DOUBLE BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8fe.gif) CMes2]NTf2 was made possible by the extreme steric shielding of the IPr** ancillary ligand (Ar=para-C6H4tBu) and the dimesitylcarbene. The strong bathochromic shift from the red-purple carbenium ion [Mes2CH]+ to the emerald-green gold–carbene cation literally shows that the carbene interaction with gold is more complex than its bonding to a proton.
CMes2]NTf2 was made possible by the extreme steric shielding of the IPr** ancillary ligand (Ar=para-C6H4tBu) and the dimesitylcarbene. The strong bathochromic shift from the red-purple carbenium ion [Mes2CH]+ to the emerald-green gold–carbene cation literally shows that the carbene interaction with gold is more complex than its bonding to a proton.
Cationic Gold Catalyst Poisoning and Reactivation

Ambient Intermolecular [2 + 2] Cycloaddition: An Example of Carbophilicity and Oxophilicity Competition in Au/Ag Catalysis

CH Bond Functionalization through Intramolecular Hydride Transfer
Abstract
Known for over a century, reactions that involve intramolecular hydride-transfer events have experienced a recent resurgence. Undoubtedly responsible for the increased interest in this research area is the realization that hydride shifts represent an attractive avenue for C![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) H bond functionalization. The redox-neutral nature of these complexity-enhancing transformations makes them ideal for sustainable reaction development. This Review summarizes recent progress in this field while highlighting key historical contributions.
H bond functionalization. The redox-neutral nature of these complexity-enhancing transformations makes them ideal for sustainable reaction development. This Review summarizes recent progress in this field while highlighting key historical contributions.

Known for over a century, reactions based on intramolecular hydride transfer have experienced a recent resurgence. Hydride shifts represent an attractive avenue for CH bond functionalization and the redox-neutral nature of these transformations makes them ideal for the development of sustainable reactions. This Review summarizes recent progress in this field and highlights key historical contributions.
[Research Article] Ligand-Controlled C(sp3)–H Arylation and Olefination in Synthesis of Unnatural Chiral α–Amino Acids
Merging Gold and Organocatalysis: A Facile Asymmetric Synthesis of Annulated Pyrroles
Abstract
The combination of cinchona-alkaloid-derived primary amine and AuI–phosphine catalysts allowed the selective CH functionalization of two adjacent carbon atoms of pyrroles under mild reaction conditions. This sequential dual activation provides seven-membered-ring-annulated pyrrole derivatives in excellent yields and enantioselectivities.

Outwitted: The combination of cinchona-alkaloid-derived primary amine and AuI–phosphine catalysts allowed the selective CH functionalization of two adjacent carbon atoms of pyrroles under mild reaction conditions. This sequential dual activation provides seven-membered-ring-annulated pyrrole derivatives in excellent yields and enantioselectivities (see scheme).
Alkyne/Alkene/Allene-Induced Disproportionation of Cationic Gold(I) Catalyst
Abstract
The first detailed experimental study of the deactivation of cationic gold was conducted, and the influence of each component in the reaction system (substrate, counterion, solvent) on the decay process was examined. It was found that a substrate (alkyne/allene/alkene)-induced disproportionation of gold(I) may play a key role in the decay process. Our mechanism is supported by kinetic, XPS, voltammetry studies, and high-resolution ESI-MS data.

The first detailed experimental study of the deactivation of cationic gold was conducted, and the influence of each component in the reaction system (substrate, counterion, solvent) on the decay process was examined. It was found that a substrate (alkyne/allene/alkene)-induced disproportionation of gold(I) may play a key role in the decay process (see scheme; TF = trifluoromethanesulfonyl).
Stereodivergent α-Allylation of Linear Aldehydes with Dual Iridium and Amine Catalysis

Silver-Catalyzed Cross-Coupling of Propargylic Alcohols with Isocyanides: An Atom-Economical Synthesis of 2,3-Allenamides
Abstract
Cross-coupling reactions between propargylic alcohols and isocyanides, by means of silver catalysis, have been described. This new reaction is both atom and step efficient and is applicable to a broad scope of substrates, allowing the synthesis of a range of synthetically valuable 2,3-allenamides in moderate to excellent yields.

Making allenamides: Cross-coupling reactions between propargylic alcohols and isocyanides, by means of silver catalysis, are described. This new reaction is both atom and step efficient and is applicable to a broad scope of substrates, allowing the synthesis of a range of synthetically valuable 2,3-allenamides in moderate to excellent yields (see scheme).
Dissecting Anion Effects in Gold(I)-Catalyzed Intermolecular Cycloadditions
Abstract
From a series of gold complexes of the type [t-BuXPhosAu(MeCN)]X (X=anion), the best results in intermolecular gold(I)-catalyzed reactions are obtained with the complex with the bulky and soft anion BAr4F− [BAr4F−=3,5-bis(trifluoromethyl)phenylborate] improving the original protocols by 10–30% yield. A kinetic study on the [2+2] cycloaddition reaction of alkynes with alkenes is consistent with an scenario in which the rate-determining step is the ligand exchange to generate the (η2-phenylacetylene)gold(I) complex. We have studied in detail the subtle differences that can be attributed to the anion in this formation, which result in a substantial decrease in the formation of unproductive σ,π-(alkyne)digold(I) complexes by destabilizing the conjugated acid formed.

Water-Soluble Gold(I) and Gold(III) Complexes with Sulfonated N-Heterocyclic Carbene Ligands: Synthesis, Characterization, and Application in the Catalytic Cycloisomerization of γ-Alkynoic Acids into Enol-Lactones

Intriguing mechanistic labyrinths in gold(I) catalysis
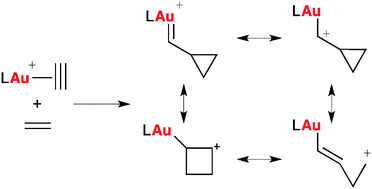
DOI: 10.1039/C3CC45518A, Feature Article
Gold(I) controls complex transformations proceeding through carbocationic species by stabilising the key reactive intermediates.
To cite this article before page numbers are assigned, use the DOI form of citation above.
The content of this RSS Feed (c) The Royal Society of Chemistry
Mechanistic Intricacies of Gold-Catalyzed Intermolecular Cycloadditions between Allenamides and Dienes
Abstract
The mechanism of the gold-catalyzed intermolecular cycloaddition between allenamides and 1,3-dienes has been explored by means of a combined experimental and computational approach. The formation of the major [4+2] cycloaddition products can be explained by invoking different pathways, the preferred ones being determined by the nature of the diene (electron neutral vs. electron rich) and the type of the gold catalyst (AuCl vs. [IPrAu]+, IPr=1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene). Therefore, in reactions catalyzed by AuCl, electron-neutral dienes favor a concerted [4+3] cycloaddition followed by a ring contraction event, whereas electron-rich dienes prefer a stepwise cationic pathway to give the same type of formal [4+2] products. On the other hand, the theoretical data suggest that by using a cationic gold catalyst, such as [IPrAuCl]/AgSbF6, the mechanism involves a direct [4+2] cycloaddition between the diene and the gold-activated allenamide. The theoretical data are also consistent with the observed regioselectivity as well as with the high selectivity towards the formation of the enamide products with a Z configuration. Finally, our data also explain the formation of the minor [2+2] products that are obtained in certain cases.

The mechanism of the gold-catalyzed intermolecular cycloaddition between allenamides and 1,3-dienes has been explored by a combined experimental and computational approach. The formation of the major [4+2] cycloaddition products can be explained by invoking different pathways, the preferred ones being determined by the nature of the diene (electron neutral vs. electron rich) and the type of gold catalyst (AuCl vs. [IPrAu]+, IPr=1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene, see scheme).