The Journal of Organic Chemistry
DOI: 10.1021/acs.joc.6b02830
 Open Access
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
A methodology for a sequential palladium-catalyzed cross-coupling procedure consisting of borylation, the Suzuki reaction and amination has been developed for the assembly of molecules with multi-aryl backbones. The linchpin of this development is the meta-terarylphosphine ligand, Cy*Phine, which has been employed as an air- and moisture-stable precatalyst, Pd(Cy*Phine)2Cl2, to improve the efficiency of one-pot borylation–Suzuki reactions. Additionally, the reactivity of the Pd-Cy*Phine system could be tuned to furnish a one-pot, borylation–Suzuki reaction–amination (BSA) cross-coupling treble. The methodology successfully integrated complementary conditions for three distinctly different and modular reactions. Average yields of 74–94% could be achieved for each segment that cumulatively afforded 50–84% yield over the entire three-step sequence in a single pot.




Traditional methods for the preparation of secondary alkyl-substituted aryl and heteroaryl chlorides challenge both selectivity and functional group tolerance. This contribution describes the use of statistical design of experiments to develop an effective procedure for the preparation of isopropyl-substituted (hetero)arenes with minimal isopropyl to n-propyl isomerization. The reaction tolerates electronically diverse aryl chloride coupling partners, with excellent conversion observed for strongly electron-deficient aromatic rings, such as esters and amides. Electron-rich systems, including methyl- and methoxy-substituted aryl chlorides, were found to be less reactive. Furthermore, the reaction was found to be most successful when heteroaryl chlorides were submitted to the cross-coupling protocol. By mapping substituent effects on reaction selectivity, we were able to show that electron-deficient aryl chlorides are essential for efficient coupling, and use electronic structure calculations to predict the likelihood of successful coupling through the estimation of the electron affinity of each aryl chloride. Moderate isolated yields were achieved with selected aryl chlorides, and moderate to good isolated yields were obtained for all the heteroaryl chlorides coupled. Excellent selectivity was observed when a 2,6-dichloroquinoline was used, allowing mono-substitution on a challenging substrate.

Halofunctionalization of alkenes is a classical method for olefin difunctionalization. It gives rise to adducts which are found in many natural products and biologically active molecules, and offers a synthetic handle for further manipulation. Classically, this reaction is performed with an electrophilic halogen source and leads to regioselective formation of the halofunctionalized adducts. Herein, we demonstrate a reversal of the native regioselectivity for alkene halofunctionalization through the use of an acridinium photooxidant in conjunction with a copper cocatalyst.

Twist of fate: Classical halofunctionalization of alkenes with an electrophilic halogen source leads to the formation of the halofunctionalized adducts with Markovnikov-type selectivity. A reversal of this native regioselectivity for alkene halofunctionalization was observed with a new dual catalytic system consisting of an acridinium photooxidant and a copper cocatalyst (see scheme; r.r.=regioisomeric ratio).


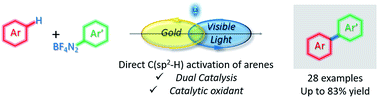
A method that allows hindered ortho-substituted aryl iodides to be efficiently coupled to phenylboronic acid using a gold-catalyzed C−C bond formation is presented. The use of a molecularly-defined dinuclear gold chloride catalytic precursor that is stabilized by a new tetradentate (N,N′)-diamino-(P,P′)-diphosphino ferrocene hybrid ligand in a Suzuki-type reaction is described for the first time. Electron-rich isopropyl groups on phosphorus were found essential for a superior activity, while the performances of a set of analogous gold dinuclear complexes that were fully characterized by multinuclear NMR spectroscopy and XRD analysis, were investigated. Therefore, arylation of para and ortho-substituted iodoarenes bearing electron-rich, electron-poor functional groups, and even hindered polycyclic aromatic compounds is described.

Gold (a)mine: Hindered ortho-substituted aryl iodides are coupled to phenylboronic acid via a gold-catalyzed C−C bond formation promoted by a novel tetradentate (N,N′)-diamino-(P,P′)-diphosphino ferrocene hybrid ligand.


Unique features of earth-abundant transition-metal catalysts are reviewed in the context of catalytic carbon–carbon bond-forming reactions. Aryl-substituted bis(imino)pyridine iron and cobalt dihalide compounds, when activated with alkyl aluminum reagents, form highly active catalysts for the polymerization of ethylene. Open-shell iron and cobalt alkyl complexes have been synthesized that serve as single-component olefin polymerization catalysts. Reduced bis(imino)pyridine iron and cobalt dinitrogen compounds have also been discovered that promote the unique [2+2] cycloaddition of unactivated terminal alkenes. Studies of the electronic structure support open-shell intermediates, a deviation from traditional strong-field organometallic compounds that promote catalytic C−C bond formation.

Homogeneous catalysis has transformed approaches to carbon–carbon bond formation. This Minireview focuses on unusual iron and cobalt complexes that promote olefin polymerization and cycloaddition reactions.



A complete switch of regioselectivity in the aminobromination of olefins is realized from delicate changes in the reaction temperature from 25 °C to 40 °C and the atmosphere from air to argon, under catalyst-free conditions. The resulting α-bromoamides can be transformed easily to high-value compounds (e.g., Clobenzorex). Mechanistic studies revealed that the reaction went through a radical pathway governed by in-situ generated bromo(tosyl)amine (TsNHBr).

The palladium(II)-catalyzed β- and γ-alkynylation of amide C(sp3)−H bonds is enabled by pyridine-based ligands. This alkynylation reaction is compatible with substrates containing α-tertiary or α-quaternary carbon centers. The β-methylene C(sp3)−H bonds of various carbocyclic rings were also successfully alkynylated.

Pyridine-enabled: The palladium(II)-catalyzed β- and γ-alkynylation of amide C(sp3)−H bonds is enabled by pyridine-based ligands. This alkynylation reaction is compatible with substrates containing α-tertiary or α-quaternary carbon centers. The β-methylene C(sp3)−H bonds in various carbocyclic rings were also successfully alkynylated.

We report herein the asymmetric coupling of flow-generated unstabilized diazo compounds and propargylated amine derivatives, using a new pyridinebis(imidazoline) ligand, a copper catalyst and base. The reaction proceeds rapidly, generating chiral allenes in 10–20 minutes with high enantioselectivity (89–98 % de/ee), moderate yields and a wide functional group tolerance.

Flow-enabled coupling: An asymmetric copper-catalyzed coupling reaction between unstabilized flow-generated diazo compounds and terminal propargylamine derivatives is described. The process occurs rapidly with high enantioselectivities, wide functional group tolerance and is further applicable to late-stage functionalization.



Nature Chemistry. doi:10.1038/nchem.2697
Authors: Jamie H. Docherty, Jingying Peng, Andrew P. Dominey & Stephen P. Thomas
NaOtBu — an alkoxide salt — enables simple access to low-oxidation-state catalysis using sustainable first-row transition metals (Fe, Co, Mn, Ni). The approach works across a wide range of reductive alkene and alkyne functionlization reactions including hydroboration, hydrosilylation, hydrogenation, hydrovinylation and [2π+2π] cyclization reactions.
Despite recent progress in the catalytic transformation of inert phenol derivatives as alternatives to aryl halides and triflates, attempts at the cross-coupling of inert phenol derivatives with the C−H bonds of arenes have met with limited success. Herein, we report the rhodium-catalyzed cross-coupling of aryl carbamates with arenes bearing a convertible directing group. The key to success is the use of an in situ generated rhodium bis(N-heterocyclic carbene) species as the catalyst, which can promote activation of the inert C(sp2)−O bond in aryl carbamates.

One for the rhodium: The rhodium-catalyzed cross-coupling of aryl carbamates with arenes bearing a convertible directing group is described. The key to success is the use of an in situ generated rhodium bis(N-heterocyclic carbene) species as the catalyst, which can promote activation of the inert C(sp2)−O bond in aryl carbamates.