Organic Letters
DOI: 10.1021/acs.orglett.6b03545

Pyridine activation by inexpensive iron catalysts has great utility, but the steps through which iron species can break the strong (105–111 kcal mol−1) C−H bonds of pyridine substrates are unknown. In this work, we report the rapid room-temperature cleavage of C−H bonds in pyridine, 4-tert-butylpyridine, and 2-phenylpyridine by an iron(I) species, to give well-characterized iron(II) products. In addition, 4-dimethylaminopyridine (DMAP) undergoes room-temperature C−N bond cleavage, which forms a dimethylamidoiron(II) complex and a pyridyl-bridged tetrairon(II) square. These facile bond-cleaving reactions are proposed to occur through intermediates having a two-electron reduced pyridine that bridges two iron centers. Thus, the redox non-innocence of the pyridine can play a key role in enabling high regioselectivity for difficult reactions.

An iron(I) complex activates C−H and C−N bonds in pyridines, using a mechanism that takes advantage of pyridine's redox activity. The bond-cleaving reactions are proposed to occur through intermediates having a two-electron reduced pyridine that bridges two iron centers.

 Open Access
Open Access   This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
Open Access
  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
The first stereoconvergent cyclopropanation reaction by means of photoredox catalysis using diiodomethane as the methylene source is described. This transformation exhibits broad functional group tolerance and it is characterized by an excellent stereocontrol en route to trans-cyclopropanes regardless of whether E- or Z-styrene substrates were utilized.

Photocatalytic approach: A stereoconvergent cyclopropanation reaction by means of photoredox catalysis using diiodomethane as the methylene source is described. This transformation exhibits broad functional group tolerance and is characterized by excellent stereocontrol en route to trans-cyclopropanes regardless of whether E- or Z-styrene substrates were utilized.
We report a robust and broadly applicable CoCl2-catalyzed cross-coupling between functionalized aryl and heteroaryl zinc pivalates and various electron-poor aryl and heteroaryl halides (X=Cl, Br, I). Couplings with (E)- or (Z)-bromo- or iodo-alkenes proceed with retention of configuration. Also, alkynyl bromides react with arylzinc pivalates providing arylated alkynes.

One pivalate to couple them all: A simple, robust, and broadly applicable cobalt salt-catalyzed procedure allows for cross-coupling reactions of functionalized aryl- and heteroarylzinc pivalates with various electron-poor aryl and heteroaryl halides (X=Cl, Br, I). Also, cross-couplings of arylzinc pivalates with bromoalkynes or alkenyl bromides and iodes proceed readily.






A stereodivergent coupling reaction between vinyl halides and boronic esters is described. This coupling process proceeds without a transition-metal catalyst, instead proceeding by electrophilic selenation or iodination of a vinyl boronate complex followed by stereospecific syn or anti elimination. Chiral, nonracemic boronic esters could be coupled with complete enantiospecificity. The process enables the highly stereoselective synthesis of either the E or Z alkene from a single isomer of a vinyl coupling partner.

Which way to go? A stereodivergent coupling process enables a highly stereoselective synthesis of alkenes, thus providing access to either isomer of the coupled product from a single isomer of a vinyl metal precursor. This coupling process proceeds without a transition-metal catalyst, instead taking place by electrophilic iodination or selenation of a vinyl boronate complex followed by stereospecific anti or syn elimination.

Open Access  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.




Nature Chemistry 8, 1126 (2016). doi:10.1038/nchem.2587
Authors: Eric C. Hansen, Dylan J. Pedro, Alexander C. Wotal, Nicholas J. Gower, Jade D. Nelson, Stephane Caron & Daniel J. Weix
Pharmaceutical compound libraries are an essential part of drug discovery and the screening of libraries for new drug leads is routine. It has now been shown that these heterocycle-rich, diverse libraries can also be used for ligand discovery. The discovery and application of several new ligands to nickel-catalysed cross-electrophile coupling is demonstrated.
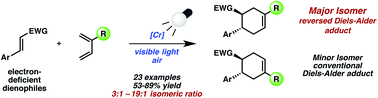
A stereoselective one-pot synthesis of functionalized complex tricyclic polyethers has been achieved using the combination of secondary amine and lanthanide catalysis. This one-pot quadruple reaction/Hetero-Diels–Alder sequence gave good yields (per step) as well as excellent diastereo- and enantioselectivities. Furthermore, the particular combination of lanthanide complexes with organocatalysis is one of the first examples described for sequential catalysis.

Targeting complexity: A unique combination of organocatalysis with lanthanide catalysis directly leads to complex tricyclic structures from readily available and simple compounds. Six new bonds and six stereocenters are formed with virtually complete stereoselectivity in this multicomponent one-pot procedure. fod=2,2-dimethyl-6,6,7,7,8,8,8-heptafluorooctane-3,5-dionato.
A novel iron-catalyzed direct alkylamination reaction of phenols has been achieved with O-benzoyl-N-alkylhydroxylamines as aminating agents. This protocol provides a facile access to N-alkyl-substituted aminophenols though a radical reaction from phenols. The catalytic direct alkylamination operates at room temperature without the need of any ligands and additives to afford the desired products with excellent regioselectivity and functional group tolerance.

A mild Heck reaction of aryl iodides and olefins was realized by the cooperation of a palladium and a photoredox catalyst under the irradiation of visible light. This protocol works well to synthesize stilbenes with high Z/E ratios and (E)-cinnamates in the absence of ligands at ambient temperature. Control experiments revealed that palladium salt, visible light and photoredox catalyst were all crucial for the achievement of this cross-coupling under mild conditions.






Versatile C−H insertions: Novel protocols for metal-carbene insertions using diazo compounds have been recently developed. Application of respective rhodium and palladium catalytic systems allow formation of new carbon–carbon bonds in very good yields and selectivities.
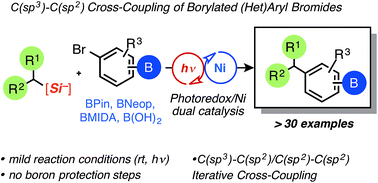
Short and highly stereoselective total syntheses of the sesquilignan natural product tatanan A and its C3 epimer are described. An assembly-line synthesis approach, using iterative lithiation–borylation reactions, was applied to install the three contiguous stereocenters with high enantio- and diastereoselectivity. One of the stereocenters was installed using a configurationally labile lithiated primary benzyl benzoate, resulting in high levels of substrate-controlled (undesired) diastereoselectivity. However, reversal of selectivity was achieved by using a novel diastereoselective Matteson homologation. Stereospecific alkynylation of a hindered secondary benzylic boronic ester enabled completion of the synthesis in a total of eight steps.

An assembly-line synthesis involving iterative lithiation-borylations enabled a short, highly enantio- and diastereoselective synthesis of tatanan A, and its C3 epimer. High substrate-controlled diastereoselectivity with a lithiated primary benzyl benzoate furnished the C3 epimer. A complete switch in selectivity was achieved using a novel substrate-controlled diastereoselective Matteson homologation to provide access to the natural product in only eight steps.