Marcos Pires
Shared posts
Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation
Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation
Nature advance online publication 04 May 2016. doi:10.1038/nature17645
Authors: Hilary P. Browne, Samuel C. Forster, Blessing O. Anonye, Nitin Kumar, B. Anne Neville, Mark D. Stares, David Goulding & Trevor D. Lawley
Our intestinal microbiota harbours a diverse bacterial community required for our health, sustenance and wellbeing. Intestinal colonization begins at birth and climaxes with the acquisition of two dominant groups of strict anaerobic bacteria belonging to the Firmicutes and Bacteroidetes phyla. Culture-independent, genomic approaches have transformed our understanding of the role of the human microbiome in health and many diseases. However, owing to the prevailing perception that our indigenous bacteria are largely recalcitrant to culture, many of their functions and phenotypes remain unknown. Here we describe a novel workflow based on targeted phenotypic culturing linked to large-scale whole-genome sequencing, phylogenetic analysis and computational modelling that demonstrates that a substantial proportion of the intestinal bacteria are culturable. Applying this approach to healthy individuals, we isolated 137 bacterial species from characterized and candidate novel families, genera and species that were archived as pure cultures. Whole-genome and metagenomic sequencing, combined with computational and phenotypic analysis, suggests that at least 50–60% of the bacterial genera from the intestinal microbiota of a healthy individual produce resilient spores, specialized for host-to-host transmission. Our approach unlocks the human intestinal microbiota for phenotypic analysis and reveals how a marked proportion of oxygen-sensitive intestinal bacteria can be transmitted between individuals, affecting microbiota heritability.
Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer
Abstract
Chimeric antigen receptor T (CAR-T) cells have demonstrated promising results against hematological malignancies, but have encountered significant challenges in translation to solid tumors. To overcome these hurdles, we have developed a switchable CAR-T cell platform in which the activity of the engineered cell is controlled by dosage of an antibody-based switch. Herein, we apply this approach to Her2-expressing breast cancers by engineering switch molecules through site-specific incorporation of FITC or grafting of a peptide neo-epitope (PNE) into the anti-Her2 antibody trastuzumab (clone 4D5). We demonstrate that both switch formats can be readily optimized to redirect CAR-T cells (specific for the corresponding FITC or PNE) to Her2-expressing tumor cells, and afford dose-titratable activation of CAR-T cells ex vivo and complete clearance of the tumor in rodent xenograft models. This strategy may facilitate the application of immunotherapy to solid tumors by affording comparable efficacy with improved safety owing to switch-based control of the CAR-T response.

CAR-T control: Chimeric antigen receptor T (CAR-T) cells were engineered to be controlled by exogenous switch molecules. Site-specific incorporation of the small molecule FITC or a short peptide neo-epitope in the anti-Her2 4D5 Fab allowed activation of corresponding switchable CAR-T cells towards Her2-expressing solid tumor cells, and displayed significant anti-cancer effects both in vitro and in vivo.
Chemical Tools To Monitor and Manipulate Adaptive Immune Responses

Self-assembly of coherently dynamic, auxetic, two-dimensional protein crystals
Nature advance online publication 02 May 2016. doi:10.1038/nature17633
Authors: Yuta Suzuki, Giovanni Cardone, David Restrepo, Pablo D. Zavattieri, Timothy S. Baker & F. Akif Tezcan
Two-dimensional (2D) crystalline materials possess unique structural, mechanical and electronic properties that make them highly attractive in many applications. Although there have been advances in preparing 2D materials that consist of one or a few atomic or molecular layers, bottom-up assembly of 2D crystalline materials remains a challenge and an active area of development. More challenging is the design of dynamic 2D lattices that can undergo large-scale motions without loss of crystallinity. Dynamic behaviour in porous three-dimensional (3D) crystalline solids has been exploited for stimuli-responsive functions and adaptive behaviour. As in such 3D materials, integrating flexibility and adaptiveness into crystalline 2D lattices would greatly broaden the functional scope of 2D materials. Here we report the self-assembly of unsupported, 2D protein lattices with precise spatial arrangements and patterns using a readily accessible design strategy. Three single- or double-point mutants of the C4-symmetric protein RhuA were designed to assemble via different modes of intermolecular interactions (single-disulfide, double-disulfide and metal-coordination) into crystalline 2D arrays. Owing to the flexibility of the single-disulfide interactions, the lattices of one of the variants (C98RhuA) are essentially defect-free and undergo substantial, but fully correlated, changes in molecular arrangement, yielding coherently dynamic 2D molecular lattices. C98RhuA lattices display a Poisson’s ratio of −1—the lowest thermodynamically possible value for an isotropic material—making them auxetic.
[Report] Conformational photoswitching of a synthetic peptide foldamer bound within a phospholipid bilayer
Diverted Total Synthesis of Promysalin Analogs Demonstrates That an Iron-Binding Motif Is Responsible for Its Narrow-Spectrum Antibacterial Activity

A bacteriophage endolysin that eliminates intracellular streptococci
PlyC, a bacteriophage-encoded endolysin, lyses Streptococcus pyogenes (Spy) on contact. Here, we demonstrate that PlyC is a potent agent for controlling intracellular Spy that often underlies refractory infections. We show that the PlyC holoenzyme, mediated by its PlyCB subunit, crosses epithelial cell membranes and clears intracellular Spy in a dose-dependent manner. Quantitative studies using model membranes establish that PlyCB interacts strongly with phosphatidylserine (PS), whereas its interaction with other lipids is weak, suggesting specificity for PS as its cellular receptor. Neutron reflection further substantiates that PlyC penetrates bilayers above a PS threshold concentration. Crystallography and docking studies identify key residues that mediate PlyCB–PS interactions, which are validated by site-directed mutagenesis. This is the first report that a native endolysin can traverse epithelial membranes, thus substantiating the potential of PlyC as an antimicrobial for Spy in the extracellular and intracellular milieu and as a scaffold for engineering other functionalities.
Rapid Bioorthogonal Chemistry Turn-on through Enzymatic or Long Wavelength Photocatalytic Activation of Tetrazine Ligation

Random peptide mixtures inhibit and eradicate methicillin-resistant Staphylococcus aureus biofilms

DOI: 10.1039/C6CC01438K, Communication
Sequence-random hydrophobic-cationic peptides are capable of controlling and managing methicillin-resistant Staphylococcus aureus biofilms and might be used as lead biofilm inhibitor candidates for further studies.
The content of this RSS Feed (c) The Royal Society of Chemistry
Mirror Images of Antimicrobial Peptides Provide Reflections on Their Functions and Amyloidogenic Properties

Extra-helical binding site of a glucagon receptor antagonist
Nature advance online publication 25 April 2016. doi:10.1038/nature17414
Authors: Ali Jazayeri, Andrew S. Doré, Daniel Lamb, Harini Krishnamurthy, Stacey M. Southall, Asma H. Baig, Andrea Bortolato, Markus Koglin, Nathan J. Robertson, James C. Errey, Stephen P. Andrews, Iryna Teobald, Alastair J. H. Brown, Robert M. Cooke, Malcolm Weir & Fiona H. Marshall
Glucagon is a 29-amino-acid peptide released from the α-cells of the islet of Langerhans, which has a key role in glucose homeostasis. Glucagon action is transduced by the class B G-protein-coupled glucagon receptor (GCGR), which is located on liver, kidney, intestinal smooth muscle, brain, adipose tissue, heart and pancreas cells, and this receptor has been considered an important drug target in the treatment of diabetes. Administration of recently identified small-molecule GCGR antagonists in patients with type 2 diabetes results in a substantial reduction of fasting and postprandial glucose concentrations. Although an X-ray structure of the transmembrane domain of the GCGR has previously been solved, the ligand (NNC0640) was not resolved. Here we report the 2.5 Å structure of human GCGR in complex with the antagonist MK-0893 (ref. 4), which is found to bind to an allosteric site outside the seven transmembrane (7TM) helical bundle in a position between TM6 and TM7 extending into the lipid bilayer. Mutagenesis of key residues identified in the X-ray structure confirms their role in the binding of MK-0893 to the receptor. The unexpected position of the binding site for MK-0893, which is structurally similar to other GCGR antagonists, suggests that glucagon activation of the receptor is prevented by restriction of the outward helical movement of TM6 required for G-protein coupling. Structural knowledge of class B receptors is limited, with only one other ligand-binding site defined—for the corticotropin-releasing hormone receptor 1 (CRF1R)—which was located deep within the 7TM bundle. We describe a completely novel allosteric binding site for class B receptors, providing an opportunity for structure-based drug design for this receptor class and furthering our understanding of the mechanisms of activation of these receptors.
Muropeptides in Pseudomonas aeruginosa and their Role as Elicitors of β-Lactam-Antibiotic Resistance
Abstract
Muropeptides are a group of bacterial natural products generated from the cell wall in the course of its turnover. These compounds are cell-wall recycling intermediates and are also involved in signaling within the bacterium. However, the identity of these signaling molecules remains elusive. The identification and characterization of 20 muropeptides from Pseudomonas aeruginosa is described. The least abundant of these metabolites is present at 100 and the most abundant at 55,000 molecules per bacterium. Analysis of these muropeptides under conditions of induction of resistance to a β-lactam antibiotic identified two signaling muropeptides (N-acetylglucosamine-1,6-anhydro-N-acetylmuramyl pentapeptide and 1,6-anhydro-N-acetylmuramyl pentapeptide). Authentic synthetic samples of these metabolites were shown to activate expression of β-lactamase in the absence of any β-lactam antibiotic, thus indicating that they serve as chemical signals in this complex biochemical pathway.

Muropeptides: A total of 20 muropeptides, which are breakdown products of peptidoglycan, were isolated from Pseuromonas aeruginosa and their structures were elucidated. The least abundant of these metabolites is present at 100 and the most abundant at 55 000 molecules per bacterium. Two of these compounds were shown to be involved in the induction of β-lactamase expression. This effect is the basis for resistance to β-lactam antibiotics in P. aeruginosa.
Polymyxins facilitate entry into mammalian cells
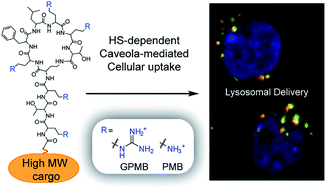
DOI: 10.1039/C6SC00488A, Edge Article
 Open Access
Open Access   This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
Polymyxin and guanidinylated polymyxin effectively deliver large biomolecules and liposomal assemblies into mammalian cells.
To cite this article before page numbers are assigned, use the DOI form of citation above.
The content of this RSS Feed (c) The Royal Society of Chemistry
Sequence-Specific 2-Cyanobenzothiazole Ligation

Nucleation and Growth of Ordered Arrays of Silver Nanoparticles on Peptide Nanofibers: Hybrid Nanostructures with Antimicrobial Properties

Inhibitory interactions promote frequent bistability among competing bacteria
Article
We know little about the effect of relationships between species on the assembly of microbial communities. Here the authors map pairwise invasion relations between bacteria and find that instead of one strain dominating, inhibitory interactions mean that often neither strain can invade the other.
Nature Communications doi: 10.1038/ncomms11274
Authors: Erik S. Wright, Kalin H. Vetsigian
Antibiotic elevates whole-genome mutagenesis [Evolution]
Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage
Nature advance online publication 20 April 2016. doi:10.1038/nature17946
Authors: Alexis C. Komor, Yongjoo B. Kim, Michael S. Packer, John A. Zuris & David R. Liu
Current genome-editing technologies introduce double-stranded (ds) DNA breaks at a target locus as the first step to gene correction. Although most genetic diseases arise from point mutations, current approaches to point mutation correction are inefficient and typically induce an abundance of random insertions and deletions (indels) at the target locus resulting from the cellular response to dsDNA breaks. Here we report the development of ‘base editing’, a new approach to genome editing that enables the direct, irreversible conversion of one target DNA base into another in a programmable manner, without requiring dsDNA backbone cleavage or a donor template. We engineered fusions of CRISPR/Cas9 and a cytidine deaminase enzyme that retain the ability to be programmed with a guide RNA, do not induce dsDNA breaks, and mediate the direct conversion of cytidine to uridine, thereby effecting a C→T (or G→A) substitution. The resulting ‘base editors’ convert cytidines within a window of approximately five nucleotides, and can efficiently correct a variety of point mutations relevant to human disease. In four transformed human and murine cell lines, second- and third-generation base editors that fuse uracil glycosylase inhibitor, and that use a Cas9 nickase targeting the non-edited strand, manipulate the cellular DNA repair response to favour desired base-editing outcomes, resulting in permanent correction of ~15–75% of total cellular DNA with minimal (typically ≤1%) indel formation. Base editing expands the scope and efficiency of genome editing of point mutations.
A Click Chemistry-Based Proteomic Approach Reveals that 1,2,4-Trioxolane and Artemisinin Antimalarials Share a Common Protein Alkylation Profile
Abstract
In spite of the recent increase in endoperoxide antimalarials under development, it remains unclear if all these chemotypes share a common mechanism of action. This is important since it will influence cross-resistance risks between the different classes. Here we investigate this proposition using novel clickable 1,2,4-trioxolane activity based protein-profiling probes (ABPPs). ABPPs with potent antimalarial activity were able to alkylate protein target(s) within the asexual erythrocytic stage of Plasmodium falciparum (3D7). Importantly, comparison of the alkylation fingerprint with that generated from an artemisinin ABPP equivalent confirms a highly conserved alkylation profile, with both endoperoxide classes targeting proteins in the glycolytic, hemoglobin degradation, antioxidant defence, protein synthesis and protein stress pathways, essential biological processes for plasmodial survival. The alkylation signatures of the two chemotypes show significant overlap (ca. 90 %) both qualitatively and semi-quantitatively, suggesting a common mechanism of action that raises concerns about potential cross-resistance liabilities.

Clickable 1,2,4-trioxolane activity-based protein-profiling probes (ABPPs) were designed to retain antimalarial activity and alkylate the molecular targets of the blood stage of Plasmodium falciparum in situ. Comparison of the 1,2,4-trioxolane protein alkylation signature with the corresponding artemisinin ABPPs indicates that both drug classes target key proteins in the glycolytic, hemoglobin degradation, antioxidant defence and protein synthesis pathways.
Real-Time Tracking and In Vivo Visualization of β-Galactosidase Activity in Colorectal Tumor with a Ratiometric Near-Infrared Fluorescent Probe

Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms
Article
Many bacteria release DNA and membrane vesicles through unclear mechanisms. Here, the authors show that a prophage endolysin is involved in the explosive lysis of a sub-population of cells in Pseudomonas aeruginosa , releasing cytoplasmic content and membrane fragments that rapidly form membrane vesicles.
Nature Communications doi: 10.1038/ncomms11220
Authors: Lynne Turnbull, Masanori Toyofuku, Amelia L. Hynen, Masaharu Kurosawa, Gabriella Pessi, Nicola K. Petty, Sarah R. Osvath, Gerardo Cárcamo-Oyarce, Erin S. Gloag, Raz Shimoni, Ulrich Omasits, Satoshi Ito, Xinhui Yap, Leigh G. Monahan, Rosalia Cavaliere, Christian H. Ahrens, Ian G. Charles, Nobuhiko Nomura, Leo Eberl, Cynthia B. Whitchurch
Bacterial-mediated phagosomal escape [Microbiology]
Nod2-mediated recognition of the microbiota is critical for mucosal adjuvant activity of cholera toxin
Nature Medicine. doi:10.1038/nm.4075
Authors: Donghyun Kim, Yun-Gi Kim, Sang-Uk Seo, Dong-Jae Kim, Nobuhiko Kamada, Dave Prescott, Dana J Philpott, Philip Rosenstiel, Naohiro Inohara & Gabriel Núñez
Plastic Antibodies for Cosmetics: Molecularly Imprinted Polymers Scavenge Precursors of Malodors
Abstract
Molecularly imprinted polymers (MIPs) are synthetic antibody mimics capable of specific molecular recognition. Advantageously, they are more stable, easy to tailor for a given application and less expensive than antibodies. These plastic antibodies are raising increasing interest and one relatively unexplored domain in which they could outplay these advantages particularly well is cosmetics. Here, we present the use of a MIP as an active ingredient of a cosmetic product, for suppressing body odors. In a dermo-cosmetic formulation, the MIP captures selectively the precursors of malodorous compounds, amidst a multitude of other molecules present in human sweat. These results pave the way to the fabrication of a novel generation of MIPs with improved selectivities in highly complex aqueous environments, and should be applicable to biotechnological and biomedical areas as well.

A new deodorant principle: Molecularly imprinted polymers (MIPs) are used for the first time as active ingredients in a cosmetic product, for suppressing body odors. In a dermo-cosmetic formulation, the MIP captures the precursors of malodorous compounds in human sweat, thus preventing them from being transformed into odorous molecules.
[Research Article] Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria
Using Chemoattractants to Lure Bacteria to Contact-Killing Surfaces
Abstract
Antimicrobial surfaces with covalently attached biocidal functionalities only kill microbes that come into direct contact with the surfaces (contact-killing surfaces). Herein, the activity of contact-killing surfaces is shown to be enhanced by using gradients in the concentration of soluble chemoattractants (CAs) to attract bacteria to the surfaces. Two natural and nonbiocidal CAs (aspartate and glucose) were used to attract bacteria to model surfaces decorated with quaternary ammonium groups (known to kill bacteria that come into contact with them). These results demonstrate the killing of Escherichia coli and Salmonella typhimurium, two common pathogens, at levels 10- to 20-times greater than that of the native surfaces alone. This approach is general and provides new strategies for the design of active or dynamic contact-killing surfaces with enhanced antimicrobial activities.

Like a moth to a flame: The activity of an antimicrobial surface coated with a biocidal agent that can kill microbes upon contact (contact-killing surface) can be enhanced substantially by using a chemoattractant (CA) concentration gradient to attract bacteria. This concept is demonstrated using two non-biocidal CAs (aspartate, glucose) to attract common foodborne bacteria to a silane-coated surface. Live bacteria=green; dead bacteria=red.
MIC score, a new tool to compare bacterial susceptibility to antibiotics application to the comparison of susceptibility to different penems of clinical strains of Pseudomonas aeruginosa
MIC score, a new tool to compare bacterial susceptibility to antibiotics application to the comparison of susceptibility to different penems of clinical strains of Pseudomonas aeruginosa
The Journal of Antibiotics advance online publication, March 30 2016. doi:10.1038/ja.2016.38
Authors: Cédric Bretonnière, Adeline Maitte, Jocelyne Caillon, Gilles Potel, David Boutoille, Cédric Jacqueline & Christophe Guitton
Direct Photocontrol of Peptidomimetics: An Alternative to Oxygen-Dependent Photodynamic Cancer Therapy
Abstract
Conventional photodynamic treatment strategies are based on the principle of activating molecular oxygen in situ by light, mediated by a photosensitizer, which leads to the generation of reactive oxygen species and thereby causes cell death. A diarylethene-derived peptidomimetic is presented that is suitable for photodynamic cancer therapy without any involvement of oxygen. This light-sensitive molecule is not a mediator but is itself the cytotoxic agent. As a derivative of the cyclic amphiphilic peptide gramicidin S, the peptidomimetic exists in two thermally stable photoforms that are interconvertible by light of different wavelengths. The isomer generated by visible light shows much stronger toxicity against tumor cells than the UV-generated isomer. First in vivo applications are demonstrated on a tumor animal model to illustrate how the peptidomimetic can be administered in the less toxic form and then activated locally in a solid tumor by visible light.

Leading light: A diarylethene-derived peptidomimetic is presented that is suitable for oxygen-independent photocontrolled cancer therapy since the light-sensitive molecule is not a mediator but the cytotoxic agent. The gramicidin S derivative exists in two thermally stable photoforms, and the isomer generated by visible light shows much stronger toxicity against tumor cells than the UV-generated isomer.
A Vancomycin Derivative with a Pyrophosphate-Binding Group: A Strategy to Combat Vancomycin-Resistant Bacteria
Abstract
Vancomycin, the drug of last resort for Gram-positive bacterial infections, has also been rendered ineffective by the emergence of resistance in such bacteria. To combat the threat of vancomycin-resistant bacteria (VRB), we report the development of a dipicolyl–vancomycin conjugate (Dipi-van), which leads to enhanced inhibition of cell-wall biosynthesis in VRB and displays in vitro activity that is more than two orders of magnitude higher than that of vancomycin. Conjugation of the dipicolyl moiety, which is a zinc-binding ligand, endowed the parent drug with the ability to bind to pyrophosphate groups of cell-wall lipids while maintaining the inherent binding affinity for pentapeptide termini of cell-wall precursors. Furthermore, no detectable resistance was observed after several serial passages, and the compound reduced the bacterial burden by a factor of 5 logs at 12 mg kg−1 in a murine model of VRB kidney infection. The findings presented in this report stress the potential of our strategy to combat VRB infections.

The glycopeptide antibiotic Dipi-van, with a zinc-binding dipicolyl moiety, is highly active against vancomycin-resistant bacteria as it binds to pyrophosphate groups of cell-wall lipids by the formation of zinc complexes while maintaining its inherent binding ability for the pentapeptide termini of cell-wall precursors. Cell-wall biosynthesis is thus inhibited.