29 Jul 14:40
Chem. Commun., 2014, 50,10645-10647
DOI: 10.1039/C4CC04366A, Communication
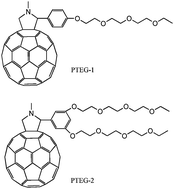
Fatemeh Jahani, Solmaz Torabi, Ryan C. Chiechi, L. Jan Anton Koster, Jan C. Hummelen
The dielectric constant of fullerene derivatives is increased through covalent modification and without deleterious effects on other properties.
The content of this RSS Feed (c) The Royal Society of Chemistry

29 Jul 06:58
by Fei Zhang, Jeanette Nangreave, Yan Liu and Hao Yan

Journal of the American Chemical Society
DOI: 10.1021/ja505101a

28 Jul 13:49
J. Mater. Chem. B, 2014, 2,6571-6579
DOI: 10.1039/C4TB01041H, Paper
Sananda Nag, Lisday Duarte, Emilie Bertrand, Veronique Celton, Mickael Castro, Veena Choudhary, Philippe Guegan, Jean-Francois Feller
A novel approach to control the selectivity of graphene-cyclodextrine sensors to cancer biomarkers by tailoring CD functionalization and assembly with graphene.
The content of this RSS Feed (c) The Royal Society of Chemistry
24 Jul 10:51
by Hong Li, Xin Li, Ying Wu, Hans Ågren and Da-Hui Qu

The Journal of Organic Chemistry
DOI: 10.1021/jo501127h

22 Jul 08:00
by Edward J. Dale, Nicolaas A. Vermeulen, Andy A. Thomas, Jonathan C. Barnes, Michal Juríček, Anthea K. Blackburn, Nathan L. Strutt, Amy A. Sarjeant, Charlotte L. Stern, Scott E. Denmark and J. Fraser Stoddart

Journal of the American Chemical Society
DOI: 10.1021/ja5041557

18 Jul 10:44
by Michael C. Young, Lauren R. Holloway, Amber M. Johnson, Richard J. Hooley
Abstract
A combination of self-complementary hydrogen bonding and metal–ligand interactions allows stereocontrol in the self-assembly of prochiral ligand scaffolds. A unique, non-tetrahedral M4L6 structure is observed upon multicomponent self-assembly of 2,7-diaminofluorenol with 2-formylpyridine and Fe(ClO4)2. The stereochemical outcome of the assembly is controlled by self-complementary hydrogen bonding between both individual ligands and a suitably sized counterion as template. This hydrogen-bonding-mediated stereoselective metal–ligand assembly allows the controlled formation of nonsymmetric discrete cage structures from previously unexploited ligand scaffolds.

Abracadabra: The stereoselective self-assembly of an unsymmetrical metal–ligand cage can be controlled by self-complementary hydrogen bonding between alcohol-containing ligands as well as between ligands and suitable anion guests.
16 Jul 14:27
Chem. Soc. Rev., 2014, Advance Article
DOI: 10.1039/C4CS00165F, Review Article
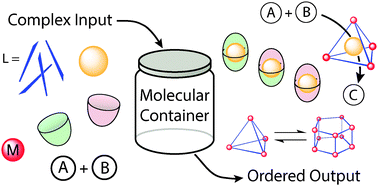
Salvatore Zarra, Daniel M. Wood, Derrick A. Roberts, Jonathan R. Nitschke
This review discusses recent advances in the use of molecular containers in complex chemical systems, focusing on three aspects: host-guest behaviour, structural transformations and reactivity modulation.
To cite this article before page numbers are assigned, use the DOI form of citation above.
The content of this RSS Feed (c) The Royal Society of Chemistry

13 Jul 16:41
J. Mater. Chem. C, 2014, 2,7239-7246
DOI: 10.1039/C4TC01210K, Paper
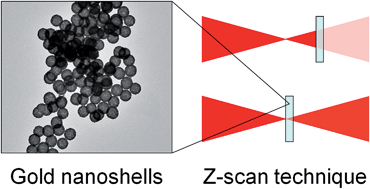
Marta Gordel, Joanna Olesiak-Banska, Radoslaw Kolkowski, Katarzyna Matczyszyn, Malcolm Buckle, Marek Samoc
Third-order nonlinear optical properties of gold nanoshells of different thickness were investigated over a broad wavelength range (530-1200 nm) by the Z-scan technique using femtosecond laser pulses.
The content of this RSS Feed (c) The Royal Society of Chemistry
10 Jul 12:36
by Ming Wang, Chao Wang, Xin-Qi Hao, Xiaohong Li, Tyler J Vaughn, Yan-Yan Zhang, Yihua Yu, Zhong-Yu Li, Mao-Ping Song, Hai-Bo Yang and Xiaopeng Li

Journal of the American Chemical Society
DOI: 10.1021/ja505414x

30 Jun 16:42
Chem. Commun., 2014, 50,9665-9668
DOI: 10.1039/C4CC03612C, Communication
Eleanor A. Wilson, Nicolaas A. Vermeulen, Paul R. McGonigal, Alyssa-Jennifer Avestro, Amy A. Sarjeant, Charlotte L. Stern, J. Fraser Stoddart
In the preparation of a hetero[3]rotaxane, the attributes of dynamic covalent chemistry are exploited by subjecting an equimolar mixture of two homo[3]rotaxanes to acid-catalysed equilibration.
The content of this RSS Feed (c) The Royal Society of Chemistry

16 Jun 11:54
by Deepak Koirala, Prakash Shrestha, Tomoko Emura, Kumi Hidaka, Shankar Mandal, Masayuki Endo, Hiroshi Sugiyama, Hanbin Mao
Abstract
While single-molecule sensing offers the ultimate detection limit, its throughput is often restricted as sensing events are carried out one at a time in most cases. 2D and 3D DNA origami nanostructures are used as expanded single-molecule platforms in a new mechanochemical sensing strategy. As a proof of concept, six sensing probes are incorporated in a 7-tile DNA origami nanoassembly, wherein binding of a target molecule to any of these probes leads to mechanochemical rearrangement of the origami nanostructure, which is monitored in real time by optical tweezers. Using these platforms, 10 pM platelet-derived growth factor (PDGF) are detected within 10 minutes, while demonstrating multiplex sensing of the PDGF and a target DNA in the same solution. By tapping into the rapid development of versatile DNA origami nanostructures, this mechanochemical platform is anticipated to offer a long sought solution for single-molecule sensing with improved throughput.

DNA origami nanostructures were used as expanded platforms for multiplex mechanochemical sensing with improved throughput at the single-molecule level. Topological rearrangements of the DNA origami nanoassemblies in response to the binding of specific targets were monitored in real time by using optical tweezers.
10 Jun 08:36
by P. Douglas Renfrew, Timothy W. Craven, Glenn L. Butterfoss, Kent Kirshenbaum and Richard Bonneau

Journal of the American Chemical Society
DOI: 10.1021/ja503776z

06 Jun 11:06
Chem. Commun., 2014, 50,11653-11656
DOI: 10.1039/C4CC03588G, Communication
Lei Wang, Weiting Yang, Yangxue Li, Zhigang Xie, Wei Zhu, Zhong-Ming Sun
A new mixed guest approach for the synthesis of heterogeneous core-shell MOF crystals was exemplified by one-pot assembly of photoactive guests into an anionic host framework.
The content of this RSS Feed (c) The Royal Society of Chemistry

05 Jun 06:52
by Jean-François Ayme, Jonathon E. Beves, Christopher J. Campbell, David A. Leigh
Abstract
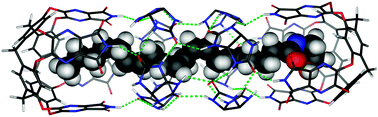
We report on multicomponent self-sorting to form open circular helicates of different sizes from a primary monoamine, FeII ions, and dialdehyde ligand strands that differ in length and structure by only two oxygen atoms. The corresponding closed circular helicates that are formed from a diamine—a molecular Solomon link and a pentafoil knot—also self-sort, but up to two of the Solomon-link-forming ligand strands can be accommodated within the pentafoil knot structure and are either incorporated or omitted depending on the stage that the components are mixed.

It takes all sorts: Tris(bidentate) ligand strands that differ in length by just two atoms self-sort into circular helicates of different sizes with a monoamine and into different molecular topologies—a molecular Solomon link and a pentafoil knot—with a diamine (see picture).
04 Jun 08:26
by Rui Zhang, Tsukasa Futagoishi, Michihisa Murata, Atsushi Wakamiya and Yasujiro Murata

Journal of the American Chemical Society
DOI: 10.1021/ja504054s

21 May 14:22
by Shaobo Ji, Wei Cao, Ying Yu, Huaping Xu
Abstract
Dynamic covalent bonds are extensively employed in dynamic combinatorial chemistry. The metathesis reaction of disulfide bonds is widely used, but requires catalysis or irradiation with ultraviolet (UV) light. It was found that diselenide bonds are dynamic covalent bonds and undergo dynamic exchange reactions under mild conditions for diselenide metathesis. This reaction is induced by irradiation with visible light and stops in the dark. The exchange is assumed to proceed through a radical mechanism, and experiments with 2,2,6,6-tetramethylpiperidin-1-yloxyl (TEMPO) support this assumption. Furthermore, the reaction can be conducted in different solvents, including protic solvents. Diselenide metathesis can also be used to synthesize diselenide-containing asymmetric block copolymers. This work thus entails the use of diselenide bonds as dynamic covalent bonds, the development of a dynamic exchange reaction under mild conditions, and an extension of selenium-related dynamic chemistry.

Diselenide bonds are dynamic covalent bonds. Their metathesis can be induced by irradiation with visible light and likely proceeds through a radical mechanism, as the exchange reaction between two different diselenides was suppressed by the addition of the radical scavenger 2,2,6,6-tetramethylpiperidine N-oxide (TEMPO).
19 May 11:47
by Miriam Peña Alvarez, Paula Mayorga Burrezo, Miklos Kertesz, Takahiro Iwamoto, Shigeru Yamago, Jianlong Xia, Ramesh Jasti, Juan T. López Navarrete, Mercedes Taravillo, Valentín G. Baonza, Juan Casado
Abstract
[n]Cycloparaphenylenes behave as molecular templates of “perfectly chemically defined” single-wall carbon nanotubes. These [n]CPP molecules have electronic, mechanical, and chemical properties in size correspondence with their giant congeners. Under mechanical stress, they form charge-transfer salts, or complexes with fullerene, by one-electron concave–convex electron transfer.

[n]Cycloparaphenylenes behave as molecular templates of “perfectly chemically defined” single-wall carbon nanotubes. These [n]CPP molecules have electronic, mechanical, and chemical properties in size correspondence with their giant congeners. Under mechanical stress, they form charge-transfer salts, or complexes with fullerene, by one-electron concave–convex electron transfer.
14 May 06:54
by Takahiro Iwamoto, Eiichi Kayahara, Nobuhiro Yasuda, Toshiyasu Suzuki, Shigeru Yamago
Abstract
A cyclic tetramer of pyrene, [4]cyclo-2,7-pyrenylene ([4]CPY), was synthesized from pyrene in six steps and 18 % overall yield by the platinum-mediated assembly of pyrene units and subsequent reductive elimination of platinum. The structures of the two key intermediates were unambiguously determined by X-ray crystallographic analysis. DFT calculations showed that the topology of the frontier orbitals in [4]CPY was essentially the same as those in [8]cycloparaphenylene ([8]CPP), and that all the pyrene units were fully conjugated. The electrochemical analyses proved the electronic properties of [4]CPY to be similar to those of [8]CPP. The results are in sharp contrast to those obtained for the corresponding linear oligomers of pyrene in which each pyrene unit was electronically isolated. The results clearly show a novel effect of the cyclic structure on cyclic π-conjugated molecules.

Changing the landscape: A cyclic tetramer of pyrene, [4]cyclo-2,7-pyrenylene ([4]CPY), was synthesized by the platinum-mediated cyclotetramerization and subsequent dehydrogenation. DFT calculations and electrochemical analyses showed that the electronic structure of [4]CPY was completely altered from that of pyrene and linear oligopyrenes. The results clearly show there is modulation of the topology of molecular orbitals upon formation of a cyclic structure.
09 May 08:15
by Andrea La Rosa, Katalin Gillemot, Edmund Leary, Charalambos Evangeli, María Teresa González, Salvatore Filippone, Gabino Rubio-Bollinger, Nicolás Agraït, Colin J. Lambert and Nazario Martín

The Journal of Organic Chemistry
DOI: 10.1021/jo500342x

05 May 11:51
by Yu Wang, Yufeng Wang, Xiaolong Zheng, Gi-Ra Yi, Stefano Sacanna, David J. Pine and Marcus Weck

Journal of the American Chemical Society
DOI: 10.1021/ja502699p

24 Apr 11:23
Chem. Soc. Rev., 2014, 43,5952-5981
DOI: 10.1039/C4CS00033A, Review Article
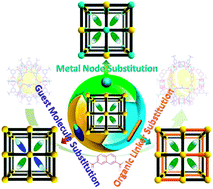
Yi Han, Jian-Rong Li, Yabo Xie, Guangsheng Guo
This review summarizes the advances in the study of substitution reactions in metal-organic frameworks (MOFs) and metal-organic polyhedra (MOPs).
The content of this RSS Feed (c) The Royal Society of Chemistry

07 Apr 13:41
Chem. Soc. Rev., 2015, 44,490-499
DOI: 10.1039/C4CS00065J, Review Article
Dariush Ajami, Lijuan Liu, Julius Rebek Jr.
The ethanolamide of arachidonic acid (anandamide) nucleates the assembly of a capsule incorporating 8 spacer units.
The content of this RSS Feed (c) The Royal Society of Chemistry

04 Apr 11:05
by Tzu-Huan Wong, Jia-Cheng Chang, Chien-Chen Lai, Yi-Hung Liu, Shie-Ming Peng and Sheng-Hsien Chiu

The Journal of Organic Chemistry
DOI: 10.1021/jo500405v

31 Mar 17:32
Chem. Soc. Rev., 2014, 43,3184-3203
DOI: 10.1039/C4CS00022F, Review Article
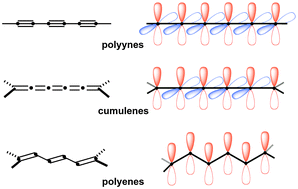
Johanna A. Januszewski, Rik R. Tykwinski
This review highlights the synthesis and reactivity of longer [n]cumulenes (n [greater-than-or-equal] 5) and summarizes their electronic and structural properties.
The content of this RSS Feed (c) The Royal Society of Chemistry

31 Mar 17:31
Chem. Soc. Rev., 2014, 43,4658-4683
DOI: 10.1039/C4CS00029C, Review Article
Nicholas H. Evans, Paul D. Beer
Key developments including the utilization of new metal cation and anion templates, dynamic combinatorial chemistry, molecular machines and the incorporation of catenanes onto surfaces and into metal organic frameworks are reviewed.
The content of this RSS Feed (c) The Royal Society of Chemistry

31 Mar 13:20
by Jean Roncali, Philippe Leriche, Philippe Blanchard
An overview of some recent developments of the chemistry of molecular donor materials for organic photovoltaics (OPV) is presented. Although molecular materials have been used for the fabrication of OPV cells from the very beginning of the field, the design of molecular donors specifically designed for OPV is a relatively recent research area. In the past few years, molecular donors have been used in both vacuum-deposited and solution-processed OPV cells and both fields have witnessed impressive progress with power conversion efficiencies crossing the symbolic limit of 10 %. However, this progress has been achieved at the price of an increasing complexity of the chemistry of active materials and of the technology of device fabrication. This evolution probably inherent to the progress of research is difficult to reconcile with the necessity for OPV to demonstrate a decisive economic advantage over existing silicon technology. In this short review various classes of molecular donors are discussed with the aim of defining possible basic molecular structures that can combine structural simplicity, low molecular weight, synthetic accessibility, scalability and that can represent possible starting points for the development of simple and cost-effective OPV materials.

Various classes of molecules used as donor materials in heterojunction organic solar cells are presented. Special emphasis is placed on molecular structures that combine low molecular weight, (in general inferior to 500) structural simplicity and synthetic accessibility with reasonable yield. Such systems are discussed as possible working structures for the development of simple and cost-effective materials for organic photovoltaics.
25 Mar 09:30
by Victor Blanco, David A. Leigh, Vanesa Marcos, José A. Morales-Serna and Alina L. Nussbaumer

Journal of the American Chemical Society
DOI: 10.1021/ja501561c

06 Mar 08:23
Publication date: September 2014
Source:Ultrasonics Sonochemistry, Volume 21, Issue 5
Author(s): Lixin Bai , Weilin Xu , Jingjun Deng , Chao Li , Delong Xu , Yandong Gao
The generation and control of acoustic cavitation structure are a prerequisite for application of cavitation in the field of ultrasonic sonochemistry and ultrasonic cleaning. The generation and control of several typical acoustic cavitation structures (conical bubble structure, smoker, acoustic Lichtenberg figure, tailing bubble structure, jet-induced bubble structures) in a 20–50kHz ultrasonic field are investigated. Cavitation bubbles tend to move along the direction of pressure drop in the region in front of radiating surface, which are the premise and the foundation of some strong acoustic cavitation structure formation. The nuclei source of above-mentioned acoustic cavitation structures is analyzed. The relationship and mutual transformation of these acoustic cavitation structures are discussed.
26 Feb 14:38
RSC Adv., 2014, 4,12133-12147
DOI: 10.1039/C4RA00615A, Paper
Nicholas G. White, Henry G. Lovett, Paul D. Beer
The use of the bis-triazolium motif to prepare potent anion receptors is described. A bis-triazolium axle containing rotaxane exhibits selective halide anion recognition in competitive solvent media.
The content of this RSS Feed (c) The Royal Society of Chemistry

07 Feb 08:18
J. Mater. Chem. A, 2014, 2,4004-4013
DOI: 10.1039/C3TA14659F, Paper
Xin Liu, QingDuan Li, Yunchuan Li, Xiong Gong, Shi-Jian Su, Yong Cao
Two indacenodithiophene core-based small molecules with different side chains were synthesized as the donor materials in organic solar cells.
The content of this RSS Feed (c) The Royal Society of Chemistry