18 May 15:32
Dalton Trans., 2016, 45,10343-10354
DOI: 10.1039/C6DT01616B, Paper
Jan Vrana, Sergey Ketkov, Roman Jambor, Ales Ruzicka, Antonin Lycka, Libor Dostal
Bis(amino)phosphanes were shown to be excellent easily accessible precursors for the preparation of a variety of tetrylenes that significantly differ in their structures.
The content of this RSS Feed (c) The Royal Society of Chemistry

18 May 10:22
by Gerald Johannes Kehr, Gerhard Erker, Stefan Grimme, Constantin Daniliuc, Xiaowu Wang, Xin Tao
Abstract
Persistent radicals undergo hydrogen atom abstraction reactions with a great variety of substrates, but not with dihydrogen. It has now been found that the TEMPO radical splits dihydrogen under mild conditions in the presence of the strong bulky B(C6F5)3 boron Lewis acid. The reaction is thought to proceed by a typical frustrated Lewis pair mechanism with the TEMPO radical acting as the active Lewis base. The reaction was analyzed by DFT, which indicates that no significant spin density on the hydrogen atoms is accumulated along the H2 splitting reaction path.

Persistent radicals undergo hydrogen atom abstraction reactions with a great variety of substrates, but not with dihydrogen. It has now been found that the TEMPO radical splits dihydrogen under mild conditions in the presence of the strong bulky B(C6F5)3 boron Lewis acid. The reaction is thought to proceed by a typical frustrated Lewis pair mechanism with the TEMPO radical acting as the active Lewis base.
17 May 10:16
Dalton Trans., 2016, 45,10355-10365
DOI: 10.1039/C6DT01154C, Paper
Xiangfei Zhang, Zexing Cao
The oxidative additions of [sigma] X-H bonds to an Al(I) center follow different mechanisms depending on their bonding features and local structural environments.
The content of this RSS Feed (c) The Royal Society of Chemistry

12 May 15:09
by Marius Schäfer, Julian Schäfer, Rian D. Dewhurst, William C. Ewing, Mirjam Krahfuß, Maximilian W. Kuntze-Fechner, Marius Wehner, Christoph Lambert, Holger Braunschweig
Abstract
The regioselective syntheses of 1,2-azaborinines is achieved using an unsymmetrical iminoborane through both catalytic and stepwise modular routes. The 1,2-azaborinine ring can be selectively functionalized in the 4- and/or 6-position through control of the stepwise reaction sequence, allowing access to vinyl-functionalized and redox-active, luminescent, donor-functionalized 1,2-azaborinines. The electrochemistry and photochemistry of a tetraarylamine-substituted 1,2-azaborinine are studied. Cyclic voltammetry of this compound, relative to a non-B,N-substituted reference molecule, showed an additional oxidation wave assigned to the oxidation of the azaborinine ring, while emission spectroscopy indicated that the azaborinine was significantly more fluorescent than the reference.

The regioselective synthesis of 1,2-azaborinines is achieved using an unsymmetrical iminoborane through both catalytic and stepwise modular routes. The 1,2-azaborinine ring can be selectively functionalized in the 2- and/or 4-position through control of the stepwise reaction sequence, allowing access to vinyl-functionalized and redox-active, luminescent, donor-functionalized 1,2-azaborinines.
12 May 14:11
by Chiemi Kojima, Ka-Ho Lee, Zhenyang Lin and Makoto Yamashita

Journal of the American Chemical Society
DOI: 10.1021/jacs.6b03686

28 Apr 13:38
by Christian P. Sindlinger, Wiebke Grahneis, Frederik S. W. Aicher, Lars Wesemann
Abstract
Hydrogen can be selectively removed from organotin trihydrides to generate the corresponding organohydrostannylene intermediates. Depending on the size of the substituent and the mode of generation, the intermediates undergo further reactions. Herein, we report on the formation of a variety of organotin hydrides with tin in the oxidation states SnII, SnI–SnIII and SnIII–SnIII, all accessed by the controlled removal of hydrogen from the tetravalent Ar′SnIV trihydride (Ar′=2,6-dimesitylphenyl, mesityl=2,4,6-trimethylphenyl).

Tin-tin time! Formation of a variety of organotin hydrides with tin in the oxidation states SnII, SnI–SnIII, and SnIII–SnIII, all accessed by the controlled removal of hydrogen from the tetravalent Ar′SnIV trihydride (Ar′=2,6-dimesitylphenyl, mesityl=2,4,6-trimethylphenyl; see scheme), is reported.
27 Apr 15:49
by Carsten Eisenhut, Nora C. Breit, Tibor Szilvási, Elisabeth Irran, Shigeyoshi Inoue
The reaction of N-heterocyclic carbene (NHC)-stabilized hydrosilylene 1, tBu3SiSi(H)![[LEFTWARDS ARROW]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/002190.gif) NHC, with one and two equivalents of benzophenone gave rise to bicyclic compounds 2 and 3, tBu3Si(R)[Si-CH2-N(CMeCMeNMe)-CPh2-O] (R = H, OCHPh2). The NHC plays a crucial role in this reactivity, as it is directly involved in C–C bond formation and supplies the C–H-activated methyl group. In the formation of 3, the terminal Si–H bond of the hydrosilylene is involved in transition-metal-free hydrosilylation. All steps in these mechanisms are based on NHC-stabilized zwitterionic transition states and intermediates and were investigated by utilizing DFT calculations. In addition, the CO2 activation of 1 to yield cis/trans-cyclotrisiloxane 4, [(tBu3Si)(H)Si-O]3, stereoselectively and its mechanistic study are reported.
NHC, with one and two equivalents of benzophenone gave rise to bicyclic compounds 2 and 3, tBu3Si(R)[Si-CH2-N(CMeCMeNMe)-CPh2-O] (R = H, OCHPh2). The NHC plays a crucial role in this reactivity, as it is directly involved in C–C bond formation and supplies the C–H-activated methyl group. In the formation of 3, the terminal Si–H bond of the hydrosilylene is involved in transition-metal-free hydrosilylation. All steps in these mechanisms are based on NHC-stabilized zwitterionic transition states and intermediates and were investigated by utilizing DFT calculations. In addition, the CO2 activation of 1 to yield cis/trans-cyclotrisiloxane 4, [(tBu3Si)(H)Si-O]3, stereoselectively and its mechanistic study are reported.

Reaction of an N-heterocyclic carbene stabilized hydrosilylene with benzophenone yields a bicyclic compound through C–C bond formation and transition-metal-free hydrosilylation. In contrast, the reaction of the same silylene with carbon dioxide affords a cis/trans-cyclotrisiloxane by oxygen transfer. Both reaction mechanisms are studied in detail by DFT calculations.
25 Apr 12:57
by Lena Albers, Judith Baumgartner, Christoph Marschner, Thomas Müller
Abstract
The ionization of 1,1-dihydridocyclopentasilane 7 has been found to yield the cyclic polysilanylsilyl cation 8 instead of the expected hydrogen-substituted silylium ion 6. The silyl cation 8 is stabilized by the formation of an intramolecular Si−H−Si bridge, which also provides the thermodynamic driving force for its formation. In general, the preference for the formation of Si−H−Si bridges can be used to scavenge and identify transient intermediates in the Lewis acid induced rearrangement of polysilanes. The validity of this concept has been demonstrated for one central step in this chemistry, the ring-contraction reaction of cyclohexasilanes to form silylcyclopentasilanes.

The hydrogen trick: A new synthetic approach has been established for trapping highly reactive and short-lived cationic intermediates of sila-Wagner–Meerwein rearrangements of polysilanes (see figure). The preference for 2e–3c Si−H−Si bridges can be used to identify transient intermediates by low-temperature 29Si NMR spectroscopy. This method allows for a general understanding of rearrangement processes in Lewis acid catalyzed polysilane transformations.
25 Apr 09:22
by Andreas Stauber, Titel Jurca, Christian Marquardt, Martin Fleischmann, Michael Seidl, George R. Whittell, Ian Manners, Manfred Scheer
A simple method to access borylphosphonium iodides [RH2P–BH2·NMe3]I (1a: R = Me; 1b: R = Et; 1c: R = nPr) by the addition of iodoalkanes to PH2–BH2·NMe3 was developed. Complexes 1a–c were characterized by multinuclear NMR spectroscopy, and 1a and 1b additionally by single-crystal X-ray diffraction. It was possible to synthesize the Lewis-base-stabilized organosubstituted phosphanylborane MePH–BH2·NMe3 (2) from [MePH2–BH2·NMe3]I (1a). Thermolysis of 2 generated a soluble, low-molecular-mass poly(alkylphosphinoborane) consisting of at least 40 repeat units, as identified by ESI-MS. These results are promising for the future preparation of a wide range of Lewis-base-stabilized phosphanylboranes, which are of interest as precursors to poly[(alkylphosphino)boranes] and are otherwise difficult to access by conventional metal-catalyzed methods.

A route for the synthesis of monoalkyl-substituted Lewis-base-stabilized phosphinoboranes via borylphosphonium precursors is presented. Mild thermolysis of the Lewis-base-stabilized methylphosphinoborane generates the corresponding polymer consisting of at least 40 repeat units, which exemplifies the applicability of this method.
21 Apr 09:40
by Eric Mädl, Gábor Balázs, Eugenia V. Peresypkina, Manfred Scheer
Abstract
The reduction of [Cp′′′Ni(η3-P3)] (1; Cp′′′=η5-1,2,4-tBu3C5H2) with potassium produces the complex anion [(Cp′′′Ni)2(μ,η2:2-P8)]2− (2), which contains a realgar-like P8 unit. The anionic triple-decker sandwich complex [(Cp′′′Ni)2(μ,η3:3-P3)]− (3) with a cyclo-P3 middle deck is obtained when 1 is treated with NaNH2 as a nucleophile. Na[3] can subsequently be oxidized with AgOTf to the neutral triple-decker complex [(Cp′′′Ni)2(μ,η3:3-P3)] (4). In contrast, 1 reacts with LiPPh2 to give the anionic compound [(Cp′′′Ni)2(μ,η2:2-P6PPh2)]− (5), a complex containing a bicyclic P7 fragment capped by two Cp′′′Ni units. Protonation of Li[5] with HBF4 leads to the neutral complex [(Cp′′′Ni)2(μ,η2:2-(HP6PPh2)] (6). Adding LiNMe2 to 1 results in [Cp′′′Ni(η2-P3NMe2)]− (7) becoming accessible, a complex which forms as a result of nucleophilic attack at the cyclo-P3 ring of 1. The complexes K2[2], Na[3], 4, 6, and Li[7] were fully characterized and their structures determined by single-crystal X-ray diffraction.

Stacking the deck: The reaction of [(η5-1,2,4-tBu3C5H2)Ni(η3-P3)] with potassium leads to the formation of a realgar-like P8 ligand. Triple-decker sandwich complexes with a P3 middle deck or unprecedented bicyclic P7 ligands are formed with nucleophiles.
19 Apr 10:43
by Matthew S. Weimer, Bo Hu, Steven J. Kraft, Roy G. Gordon, Carlo U. Segre and Adam S. Hock

Organometallics
DOI: 10.1021/acs.organomet.5b01004

19 Apr 10:42
by Felix S. Geitner, Thomas F. Fässler
Reaction of the functionalized tetrel cluster [Ge9R3]– {R = Si(SiMe3)3} with coinage metal N-heterocyclic carbene (NHC) complexes affords the first Zintl cluster transition metal complexes coordinated by NHC molecules. In [(η3-Ge9R3)M(NHCDipp)], the D3d-symmetric cluster polyhedron coordinates with a triangular Ge3 face to the coinage metals M = Cu, Ag, Au, which themselves also bind to the NHC. The role of fine tuning of electronic properties by using an NHC ligand is discussed for the transformation reaction of [(η3-Ge9R3)Ag(NHCDipp)] to yield [Ag(η3-Ge9R3)2][Ag(NHCDipp)2]. All complexes are fully characterized including single-crystal X-ray structure analyses.

Whereas intermetalloid cluster formation from ligand-free Zintl ions affords highly polar solvents, functionalized Zintl cluster anions allow for other solvents. Reactions with coinage metal N-heterocyclic carbene (NHC) complexes give a deep insight into the reactivity of Zintl clusters, as shown by the series [(η3-Ge9R3)M(NHCDipp)], M = Cu, Ag, Au and the subsequent transformation to [M(η3-Ge9R3)2][M(NHCDipp)2] for M = Ag.
12 Apr 13:22
Dalton Trans., 2016, 45,7226-7230
DOI: 10.1039/C6DT01015F, Communication
Nada Y. Tashkandi, Laura C. Pavelka, Christine A. Caputo, Paul D. Boyle, Philip P. Power, Kim M. Baines
The reaction pathway for the addition of alkynes to digermynes has been investigated using a mechanistic probe approach.
The content of this RSS Feed (c) The Royal Society of Chemistry

04 Apr 13:03
Dalton Trans., 2016, 45,6044-6052
DOI: 10.1039/C5DT02711J, Paper

Open Access
Alexander Hinz, Julia Rothe, Axel Schulz, Alexander Villinger
A bulky NPN-substituted dichlorostibane was reduced with KC8 to afford a distibenium compound with a [Sb2]2+ ion which can be regarded as a dimerization product of a stiba-phospha-diazanediyl singlet biradicaloid. The intermediate formation of this singlet stiba-phospha-diazanediyl was proven by trapping experiments.
The content of this RSS Feed (c) The Royal Society of Chemistry

04 Apr 12:52
by Baojie Feng

Nature Chemistry.
doi:10.1038/nchem.2491
Authors: Baojie Feng, Jin Zhang, Qing Zhong, Wenbin Li, Shuai Li, Hui Li, Peng Cheng, Sheng Meng, Lan Chen & Kehui Wu
A variety of two-dimensional materials have been reported in the past few years, yet single-element systems—such as graphene and black phosphorus—have remained rare. 2D allotropes of boron have long been predicted and recently investigated. Two boron sheets have now been grown on a Ag(111) surface by molecule beam epitaxy that exhibit significant chemical stability against oxidation.
04 Apr 11:24
by Andrey V. Protchenko, Joshua I. Bates, Liban M. A. Saleh, Matthew P. Blake, Andrew D. Schwarz, Eugene L. Kolychev, Amber L. Thompson, Cameron Jones, Philip Mountford and Simon Aldridge

Journal of the American Chemical Society
DOI: 10.1021/jacs.6b00710

04 Apr 11:21
by Conor Pranckevicius, Liu Liu, Guy Bertrand, Douglas W. Stephan
Abstract
The first carbodicarbene stabilized by flanking cyclopropenylidenes is reported. Tetraphenylcarbodicyclopropenylidene (2) is accessed by deprotonation of the corresponding triafulvene cyclopropenium salt, and has been spectroscopically characterized in [D8]THF solution at −60 °C. Main-group and transition-metal complexes of 2 have been accessed, and have revealed the high sigma donating ability, and exclusive η1 binding of this neutral all carbon ligand. Variable temperature NMR spectroscopy studies reveal varying degrees of free rotation in the flanking cyclopropenylidene groups of 2 in its coordination compounds.

Carbo loading: The first carbodicarbene stabilized by flanking cyclopropenylidenes is accessed by deprotonation of the corresponding triafulvene cyclopropenium salt. Main-group and transition-metal complexes of it have been accessed, and have revealed the high σ-donating ability, and exclusive η1 binding of this neutral all carbon ligand.
28 Mar 13:18
by Guo-Qiang Chen, Gerald Kehr, Constantin G. Daniliuc, Christian Mück-Lichtenfeld, Gerhard Erker
Abstract
The phosphorus/boron-substituted hexatriene systems 6 undergo thermally induced electrocyclic ring closure to yield the cyclohexadiene-derived P/B frustrated Lewis pairs (FLPs) 7. Subsequent TEMPO oxidation gives the phenylene-bridged FLPs 8. Both systems activate dihydrogen and the thermally robust FLPs undergo carbon–carbon coupling reactions at a mesityl group upon treatment with dimethyl acetylenedicarboxylate at elevated temperatures.

The phosphorus/boron-substituted hexatriene gave the phenylene-bridged P/B frustrated Lewis pairs (FLP) by thermally induced electrocyclic ring closure followed by oxidation with the 2,2,6,6-tetramethylpiperidine-N-oxyl radical. The thermally robust FLP reacted with dimethyl acetylenedicarboxylate by C−C coupling with the mesityl group (see picture).
21 Mar 14:46
by Iva Vránová, Mercedes Alonso, Roman Jambor, Aleš Růžička, Milan Erben, Libor Dostál
Abstract
The reaction of stibinidene and bismuthinidene ArM [where Ar=C6H3-2,6-(CH=NtBu)2; M=Sb (1), Bi (2)] with transition metal (TM) carbonyls Co2(CO)8 and Mn2(CO)10 produced unprecedented ionic complexes [(ArM)2Co(CO)3]+[Co(CO)4]− and [(ArM)2Mn(CO)4]+[Mn(CO)5]− [where M=Sb (3, 5), Bi (4, 6)]. The pnictinidenes 1 and 2 behaved as two-electron donors in this set of compounds. Besides the M![[RIGHTWARDS ARROW]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/002192.gif) TM bonds, the topological analysis also revealed a number of secondary interactions contributing to the stabilization of cationic parts of titled complexes.
TM bonds, the topological analysis also revealed a number of secondary interactions contributing to the stabilization of cationic parts of titled complexes.

Generous donation: The synthesis, structure and bonding analysis of stibinidene and bismuthinidene ionic complexes of cobalt and manganese is presented. The pnictinidenes are coordinated as terminal ligands and act as two-electron donors in this set of compounds, an unprecedented feature in the chemistry of heavy Group 15 elements.
18 Mar 09:43
by Rian D. Dewhurst, Ralph Claessen, Holger Braunschweig

The construction of single-atom-thick sheets of boron on a silver substrate, which was published in late 2015, represents a significant advance towards the realization of useful two-dimensional materials based solely on boron. This Highlight provides background information on the topic of boron allotropes and an outlook for further work in this area.
09 Mar 09:39
by Natalia Del Rio, Antoine Baceiredo, Nathalie Saffon-Merceron, Daisuke Hashizume, Dennis Lutters, Thomas Müller, Tsuyoshi Kato
Abstract
A new stable heterocyclic germylene, in which the divalent germanium atom lies between a nitrogen atom and a phosphanylidene phosphorane group, was synthesized. Experimental and theoretical studies revealed the peculiar effect of phosphanylidene phosphorane substituent, which is a stronger π-donor towards germanium than an amino group is. Because of the weak phosphorus–germanium π-bond, this new germylene compound shows an enhanced reactivity compared to classical N-heterocyclic germylenes.

‘Germs’ of a different type: A stable heterocyclic germylene, in which the divalent germanium atom lies between a nitrogen atom and a phosphanylidene phosphorane group, was synthesized. Experimental and theoretical studies revealed the peculiar effect of the phosphanylidene phosphorane substituent, which presents π-donor abilities stronger than that of amino groups.
08 Mar 17:05
by Suresh Mummadi, Daniel K. Unruh, Jiyang Zhao, Shuhua Li and Clemens Krempner

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b13545

29 Feb 13:26
by Sebastian Litters, Elisabeth Kaifer, Hans-Jörg Himmel
Abstract
The red-colored tetraborane(4) [B4(hpp)4]3+. (3; hpp=1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]pyrimidinate) with a rhomboid B4 skeleton stabilized by four N donors, was synthesized by the reaction of the strong hydride abstraction reagent [(acridine)BCl2][AlCl4] with the electron-rich diborane(4) [HB(hpp)]2 (1). The salt 3[AlCl4]3 was structurally characterized and the presence of unpaired electrons proven by EPR measurements. The unprecedented radical tricationic 3 is distinguished by a high positive charge and boron atoms in a low oxidation state (less than two).

Share and share alike: Reaction of the doubly base-stabilized diborane(4) [HB(hpp)]2 with the strong hydride abstraction reagent [(acridine)BCl2][AlCl4] leads to the unprecedented red-colored tetraborane(4) [B4(hpp)4]3+. with a rhomboid B4 skeleton and four-center, five-electron bonding.
26 Feb 09:40
by Michael J Cowley, Amy N Price
Abstract
We report the preparation of N-heterocyclic carbene (NHC)-stabilized compounds containing P=B double bonds. The reaction of the highly functionalized phosphinoborane Mes*(SiMe3)P−B(Cl)Cp* with Lewis bases allows access to base-stabilized phosphinidene boranes Mes*P=B(L)Cp* (L=4-dimethylaminopyridine (DMAP), NHC) by Me3SiCl elimination. The formation of these species is shown to proceed through transient borylphosphide anions generated by Me3Si abstraction.

Intermediate intercepted! Syntheses of a range of base-stabilized P=B doubly-bonded compounds proceeds by SiMe3 abstraction (see scheme; Mes*=2,4,6-tritertbutylphenyl; Cp*=1,2,3,4,5-pentamethylcyclopentadienyl). The interception of an anionic borylphosphide intermediate offers insight into thermal Me3SiCl elimination, for example in chemical vapor deposition processes.
12 Feb 16:08
Chem. Sci., 2016, 7,3384-3389
DOI: 10.1039/C5SC04798F, Edge Article
Open Access
John S. McGough, Samuel M. Butler, Ian A. Cade, Michael J. Ingleson
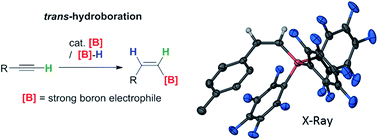
Transition metal free alkyne trans-hydroboration is achieved using strong boron electrophiles in the presence of a B-H moiety.
The content of this RSS Feed (c) The Royal Society of Chemistry

10 Feb 11:33
by Chandrajeet Mohapatra, Prinson P. Samuel, Bin Li, Benedikt Niepötter, Christian J. Schürmann, Regine Herbst-Irmer, Dietmar Stalke, Bholanath Maity, Debasis Koley and Herbert W. Roesky

Inorganic Chemistry
DOI: 10.1021/acs.inorgchem.6b00024

05 Feb 10:34
by Florenz Buß, Paul Mehlmann, Christian Mück-Lichtenfeld, Klaus Bergander and Fabian Dielmann

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b13116

03 Feb 09:43
by Rahul Kumar Siwatch, Surendar Karwasara, Mahendra Kumar Sharma, Santigopal Mondal, Goutam Mukherjee, Gopalan Rajaraman and Selvarajan Nagendran

Organometallics
DOI: 10.1021/acs.organomet.5b00643

02 Feb 12:54
Dalton Trans., 2016, 45,9820-9826
DOI: 10.1039/C6DT00300A, Paper
Anthony D. Ledet, Todd W. Hudnall
We have synthesized the first diamidocarbene (DAC)-supported borenium salt which was found to readily undergo two sequential 1-electron reductions to give a boryl-substituted DAC-centred radical and a DAC-supported aminoborylene.
The content of this RSS Feed (c) The Royal Society of Chemistry

02 Feb 12:53
by Kamil Samigullin, Isabelle Georg, Michael Bolte, Hans-Wolfram Lerner, Matthias Wagner
Abstract
The geminal frustrated Lewis pair tBu2PCH2B(Fxyl)2 (1; Fxyl=3,5-(CF3)2C6H3) is accessible in 65 % yield from tBu2PCH2Li and (Fxyl)2BF. According to NMR spectroscopy and X-ray crystallography, 1 is monomeric both in solution and in the solid state. The intramolecular P⋅⋅⋅B distance of 2.900(5) Å and the full planarity of the borane site exclude any significant P/B interaction. Compound 1 readily activates a broad variety of substrates including H2, EtMe2SiH, CO2/CS2, Ph2CO, and H3CCN. Terminal alkynes react with heterolysis of the C−H bond. Haloboranes give cyclic adducts with strong P−BX3 and weak R3B−X bonds. Unprecedented transformations leading to zwitterionic XP/BCX3 adducts occur on treatment of 1 with CCl4 or CBr4 in Et2O. In less polar solvents (C6H6, n-pentane), XP/BCX3 adduct formation is accompanied by the generation of significant amounts of XP/BX adducts. FLP 1 catalyzes the hydrogenation of PhCH=NtBu and the hydrosilylation of Ph2CO with EtMe2SiH.

Complementary twins: The geminal frustrated Lewis pair tBu2PCH2B[3,5-(CF3)2C6H3]2 (1) is accessible from tBu2PCH2Li and [3,5-(CF3)2C6H3]2BF. Compound 1 readily activates element–element single, double, and triple bonds. Remarkably, CX4 undergoes C−X bond heterolysis with formation of unprecedented XP/BCX3 adducts (X=Cl, Br; see scheme).