Publication date: 15 June 2021
Source: Coordination Chemistry Reviews, Volume 437
Author(s): Zhen Ma, Kamran T. Mahmudov, Vusala A. Aliyeva, Atash V. Gurbanov, M. Fátima C. Guedes da Silva, Armando J.L. Pombeiro
Publication date: 15 June 2021
Source: Coordination Chemistry Reviews, Volume 437
Author(s): Zhen Ma, Kamran T. Mahmudov, Vusala A. Aliyeva, Atash V. Gurbanov, M. Fátima C. Guedes da Silva, Armando J.L. Pombeiro
Nature Reviews Drug Discovery, Published online: 09 March 2021; doi:10.1038/d41573-021-00041-7
New target for multiple myelomaNature Reviews Drug Discovery, Published online: 16 March 2021; doi:10.1038/d41573-021-00050-6
Top companies and drugs by sales in 2020




Catalytic C−H methylation proceeds efficiently under solvent‐free, mechanochemical conditions, including the late‐stage functionalization of biologically active substrates and otherwise difficult‐to‐make metalacyclic complexes.
The mechanochemical, solvent‐free, highly regioselective, rhodium‐catalyzed C−H methylation of (hetero)arenes is reported. The reaction shows excellent functional‐group compatibility and is demonstrated to work for the late‐stage C−H methylation of biologically active compounds. The method requires no external heating and benefits from considerably shorter reaction times than previous solution‐based C−H methylation protocols. Additionally, the mechanochemical approach is shown to enable the efficient synthesis of organometallic complexes that are difficult to generate conventionally.


Global switch for local effect: The linear fusion of (aza)acene and a redox switchable macrocycle (i.e. triphyrin(2.1.1)) gives a set of fused systems merging two π‐conjugated clouds. A competition between local and global effects can be distinguished as documented in spectroscopic properties with a domination of (aza)acene diatropic current obtained after a redox switching within the macrocyclic flank.
A strong conjugation present in fused systems plays a crucial role in tuning of the properties that would be showing a dependence on the efficiency of π‐electrons coupling. The π‐cloud available in the final structure can be drastically influenced by a side‐ or a linear fusion of unsaturated and conjugated hydrocarbons. The linear welding of naphthalene/anthracene or quinoxaline/benzo[g]quinoxaline with triphyrin(2.1.1) gives structures where the competition between local and global delocalization is distinguished. The aromatic character observed in skeletons strongly depends on the oxidation state of the macrocyclic flanking and is either extended over the whole system or kept as a composition of local currents (diatropic and paratropic) of incorporated units. The hybrid systems show the properties derived from the π‐conjugations that interlace one another but also show a significant independence of (aza)acene subunits reflected in the observed spectroscopic properties.

Isocyanide‐based reactions: The development of catalytic enantioselective isocyanide‐based reactions is currently of great interest because the resulting products are valuable in organic synthesis, pharmacological chemistry, and materials science. This review assembles and comprehensively summarizes the recent achievements in this rapidly growing area according to the reaction types. Special attention is paid to the advantages, limitations, possible mechanisms, and synthetic applications of each reaction. In addition, a personal outlook on the opportunities for further exploration is given.
The development of catalytic enantioselective isocyanide‐based reactions is currently of great interest because the resulting products are valuable in organic synthesis, pharmacological chemistry, and materials science. This review assembles and comprehensively summarizes the recent achievements in this rapidly growing area according to the reaction types. Special attention is paid to the advantages, limitations, possible mechanisms, and synthetic applications of each reaction. In addition, a personal outlook on the opportunities for further exploration is given at the end.

The future of Ca: A novel, sustainable, modular and high yielding synthesis of medicinally important scaffolds has been realized. The reaction is tolerant of a range of functional groups and produces environmentally benign alcoholic by‐products. The rapid nature of the reaction, coupled with the modulatory of the methodology, means it should find use in a range of applications.
Herein, we report a sustainable, modular, rapid and high‐yielding transformation to afford densely functionalized 5‐aminooxazoles and thiazoles. The reaction is tolerant to a wide range of functional groups and is typically complete in under 30 min. Furthermore, the described transformation is inherently green in relation to the catalyst and solvent choice as well as producing environmentally benign alcoholic by‐products.


This Concept article discusses recent applications of commercial organic superbases to new synthetic methodology highlighting their advantages compared to common inorganic bases in the context of (1) new base‐catalyzed reactions, (2) methods that employ stoichiometric bases, and (3) high‐throughput experimentation.
Organic superbases are a distinct and increasingly utilized class of Brønsted base that possess properties complementary to common inorganic bases. This Concept article discusses recent applications of commercial organic superbases in modern synthetic methodologies. Examples of the advantages of organic superbases in three areas are highlighted, including the discovery of new base‐catalyzed reactions, the optimization of reactions that require stoichiometric Brønsted base, and in high‐throughput experimentation technology.

Vacuum‐field strong coupling with molecular vibrations imposes symmetry constraints which alter the Woodward–Hoffmann selectivity of cyclobutene ring opening under thermal conditions.
Vibrational strong coupling (VSC) has recently been shown to change the rate and chemoselectivity of ground‐state chemical reactions via the formation of light–matter hybrid polaritonic states. However, the observation that vibrational‐mode symmetry has a large influence on charge‐transfer reactions under VSC suggests that symmetry considerations could be used to control other types of chemical selectivity through VSC. Here, we show that VSC influences the stereoselectivity of the thermal electrocyclic ring opening of a cyclobutene derivative, a reaction which follows the Woodward–Hoffmann rules. The direction of the change in stereoselectivity depends on the vibrational mode that is coupled, as do changes in rate and reaction thermodynamics. These results on pericyclic reactions confirm that symmetry plays a key role in chemistry under VSC.

Synlett
DOI: 10.1055/a-1389-9498

Cyclophanes are an admirable class of macrocyclic and cage compounds that often display unusual properties due to their high strain and unusual conformations. However, the exploration of new, complex cyclophanes has been encumbered by syntheses that can be low yielding, require harsh reaction conditions, and arduous purification steps. Herein, we discuss our work using metalloid-directed self-assembly and dynamic covalent chemistry to form cryptands. These were then subjected to mild conditions to produce discrete disulfide, thioether and hydrocarbon macrocycles in high yields. ‘Design of Experiments’ was then used to selectively synthesize targeted macrocycles from complex mixtures.1 Introduction2 Cryptands to Cyclophanes3 Functionalizable Macrocycles4 ‘Design of Experiments’ Targeted Synthesis5 Conclusions and Outlook
[...]
Georg Thieme Verlag KG Rüdigerstraße 14, 70469 Stuttgart, Germany
Article in Thieme eJournals:
Table of contents | Abstract | Full text

A comprehensive study of photoisomerzation of ferulic acid derived esters, amides, and ketones was carried out. Only in the case of aliphatic tertiary amides, a nearly complete E→Z conversion was achieved.
A thorough study on the (E) to (Z) photoisomerization of ferulic acid derivatives (esters, amides of all types, and ketones) was carried out. At the photostationary state, only aliphatic or benzylic tertiary amides reach a nearly complete conversion of (E) isomers into the (Z) ones, whereas for esters, primary and secondary amides or aromatic tertiary amides mixtures of (Z)/(E) ranging from 7 : 93 to 72 : 28 are observed. Ketones show rather limited photoisomerization. However, (Z) ketones may be obtained by the reaction of organometal compounds with an isomerized (Z) Weinreb amide.

From basics to advanced: Chirality in triptycenes due to atropisomerism and stereocenters have been reviewed and the routes to obtain these chiral triptycenes have been highlighted. This Minireview describes the initial history of developments of chiral triptycenes and its comeback to important applications in advanced materials and catalysis after a gap of almost 30 years.
Triptycenes have been established as unique scaffolds because of their backbone π‐structure with a propeller‐like shape and saddle‐like cavities. They are some of the key organic molecules that have been extensively studied in polymer chemistry, in supramolecular chemistry and in material science. Triptycenes become chiral molecules when substituents are unsymmetrically attached in at least two of their different aromatic rings. This Minireview highlights the chirality of triptycenes from basics to an advanced stage for the development of functional molecules.


The introduction of trifluoromethylchalcogen group (CF3O, CF3S and CF3Se) has attracted growing attention in the field of modern organofluorine chemistry. Compared with the CF3S and CF3O chemistry, methods for trifluoromethylselenolation are much less developed owing to the limited availability of CF3Se transfer reagents and synthetic methods. The CF3Se group introduces promising lipophilicity (Hansch‐Leo parameter=1.29) and strong electron‐withdrawing effect (Hammett constants σm=0.44, σp=0.45) which are important parameters for the development of new pharmaceutically relevant compounds. Traditionally, the CF3Se compounds were synthesized by the nucleophilic trifluoromethylation of diselenides and selenocyanates, which suffered from harsh conditions and/or limited substrates scope. Compared with the indirect methods, the direct formation of C−SeCF3 constitutes is a more efficient approach. In recent years, new reagents and methods were developed which enabled the incorporation of the trifluoromethylselenyl group directly under mild conditions, specifically in transition metal catalysis and photoredox catalysis. In this review, we will focus on direct trifluoromethylselenolation strategies based on the development of new trifluoromethylselenolation reagents and methods.

This Review summarizes advances in photoredox-mediated Giese reactions since 2013, with a focus on the breadth of methods that provide access to crucial carbon-centered radical intermediates that can engage in radical conjugate addition processes.
Photomediated Giese reactions are at the forefront of radical chemistry, much like the classical tin-mediated Giese reactions were nearly forty years ago. With the global recognition of organometallic photocatalysts for the mild and tunable generation of carbon-centered radicals, chemists have developed a torrent of strategies to form previously inaccessible radical intermediates that are capable of engaging in intermolecular conjugate addition reactions. This Review summarizes advances in photoredox-mediated Giese reactions since 2013, with a focus on the breadth of methods that provide access to crucial carbon-centered radical intermediates that can engage in radical conjugate addition processes.
Abstract
The rhodium-catalyzed transannulation of N-perfluoroalkyl-1,2,3-triazoles with aromatic and aliphatic terminal alkynes under microwave heating conditions afforded N-perfluoroalkyl-3,4-disubstituted pyrroles (major products) and N-fluoroalkyl-2,4-disubstituted pyrroles (minor products). The observed selectivities in the case of the reactions with aliphatic alkynes were high.
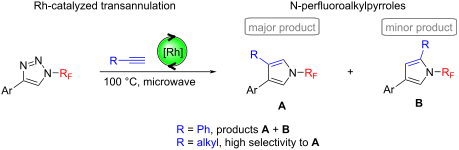
Beilstein J. Org. Chem. 2021, 17, 504–510. doi:10.3762/bjoc.17.44
Abstract
The difunctionalization of alkenes involving a trifluoromethylthio group (SCF3) for the conversion of versatile and readily available olefins into structurally more complex molecules has been successfully studied. However, the disproportionate dithiolation of alkenes is unknown. Herein, a transition-metal-free protocol is presented for the vicinal trifluoromethylthio–thiolation of unactivated alkenes via a radical process under mild conditions with a broad substrate scope and excellent tolerance.
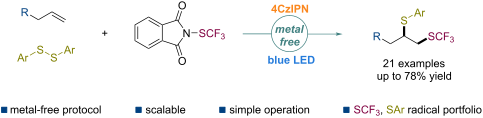
Beilstein J. Org. Chem. 2021, 17, 551–557. doi:10.3762/bjoc.17.49

The visible‐light‐induced intramolecular double dearomative cycloaddition of arenes is reported. The reactions are carried out under mild conditions and feature a wide substrate scope. A large array of polycyclic indoline derivatives is afforded via dearomative [4+2] or [2+2] cycloaddition pathways in high yields with exclusive diastereoselectivity.
Herein we report visible‐light‐induced intramolecular double dearomative cycloaddition of arenes. Compared with the well‐known photodimerization of arenes under ultraviolet irradiation, the current reactions are carried out under mild conditions and feature wide substrate scope. A large array of structurally‐diverse polycyclic indoline derivatives is afforded in high yields (up to 98 %) with exclusive diastereoselectivity (>20:1 dr) via dearomative [4+2] or [2+2] pathway.


