This beautiful little boy has heritable retinoblastoma. The white spots in his eyes are from light reflecting off of tumors.
In a list of famous genes, RB1 would probably be #1. It’s the tumor suppressor gene whose “loss of function” is behind the childhood eye cancer retinoblastoma, and that Alfred Knudson investigated to deduce the 2-hit mechanism of cancer.
In 1971, the idea that a gene’s normal function could be to prevent cancer was revolutionary. Now a new study finds that an amplified oncogene can cause the eye cancer too — with just one hit.
TWO ROUTES TO A TUMOR
Like the earth being flat, proteins being the genetic material, and genes being contiguous, the idea that mutations in RB1 are the sole cause of retinoblastoma has vanished. A multinational team of researchers, led by Brenda Gallie of Impact Genetics and the Toronto Western Hospital Research Institute, discovered that in a very small proportion of children with retinoblastoma, an oncogene, MYCN, is the cause. Their report is in Lancet Oncology. (Dr. Gallie, who supplied the photos here, heads an international effort to bring care for RB to the 92% of the thousands of children who have the disease who live in developing countries.)

Untreated retinoblastoma, circa 1806.
Retinoblastoma has a long history. A 2000 B.C. Mayan stone carving shows a child with a bulging eye. A Dutch anatomist provided the earliest clinical description, a growth “the size of two fists.” In 1886 researchers noted that the cancer can be inherited, and in those families, secondary tumors can arise, usually in bone. Once flash photography was invented, parents would notice the disease as white spots in the pupil, from light reflecting off a tumor.
About 1 in 20,000 infants has RB. In 40% of cases, both eyes develop tumors. This means that the child inherited a predisposition mutation in all cells, and the cancer develops when a second mutation “hits” a retinal cell. Of the 60% of cases where only one eye is affected, most (85%) result from 2 hits in the same retinal cell, without the inherited (aka germline or constitutional) mutation.
The road to finding the new gene began when the investigators in Toronto noticed that 7 children who had a tumor in just one eye didn’t have the expected RB1 mutation. Instead, one of the genes that typically comes into play later in the disease is present in extra copies – the oncogene MYCN. It’s known to cause neuroblastoma, another cancer that begins so early that babies are born with it. Just one amplified MYCN, like a genetic stutter, is sufficient to trigger the eye cancer. In genetics lingo, it’s a dominant, “gain-of-function” mutation.

Rosette structures are seen in classic RB tumors, but not in the newly discovered kind.
Researchers from New Zealand, France, Germany, and the Netherlands added cases, for a total of 1068, that included more children with the newly-recognized form of the disease, dubbed MYCN RB. The MYCN tumor itself, for those who like to look at such things, appears different. The rosette-like structures and coalesced nuclei seen in classic RB are absent, and the cells look like those of neuroblastoma. “It’s a subtle enough distinction that pathologists who received an eye removed under suspicion of RB would be excused if they say it looks like RB,” Timothy Corson PhD, assistant professor of ophthalmology, biochemistry and molecular biology at the Indiana University School of Medicine, told me.
The tumors of MYCN RB are large, invasive, and so aggressive that they usually appear before a child is six months old. In fact, 18% of one-eye retinoblastomas in kids this young are of the new type.
CLINICAL IMPLICATIONS
The finding of a new form of retinoblastoma has implications both specific and general.
Knowledge improves the lives of patients and their families. “If an infant with a single tumor is diagnosed with MYCN RB, then it’s not heritable. These kids don’t have the risk to pass it on to offspring, nor do they face a greater risk for subsequent cancers,” Corson said. That’s because the new form of RB arises anew in one gene in a retinal cell, not in sperm or eggs.
Follow-up is easier. “The exams through childhood are not simply going to an ophthalmologist and he or she looks in the eye. An infant must be put under general anesthesia to be still enough to carefully evaluate the eyes for tumors,” Corson says.
Treatment changes too. The first eye of a child with a family history of the disease is usually treated with chemo, rather than being removed, because the second eye is likely to develop tumors too. Preserving the eye saves sight. But in MYCN RB, “since the disease is particularly aggressive, and the other eye will not become involved, surgical removal of the diseased eye is the correct therapeutic course,” says Joan O’Brien, MD, chair of ophthalmology at the University of Pennsylvania School of Medicine.

A retinoblastoma tumor grows on the retina.
In addition to RB1 being a famous gene, the new finding enhances what is already a medical success story. “In ophthalmology we don’t often deal in life and death issues. Retinoblastoma is uniformly fatal if not treated. It’s wonderful that most of these young kids are cured nowadays,”shares Shalom Kieval, MD, associate clinical professor of ophthalmology, Albany Medical College, president of RetinaCare Consultants, Albany, New York, and chief of ophthalmology at Albany Memorial Hospital. One of his patients, whom he first met as a premature infant, brings her three kids to see him now. The mother and two of her kids have been successfully treated for RB. “The retinoblastoma story is another unsung but intensely gratifying accomplishment of modern medical science,” he adds.
THE BIGGER PICTURE
“Genetic heterogeneity” is when more than one genotype (gene variant combo) is responsible for the same phenotype (disease or trait). Hearing loss, for example, has more than 100 different genetic causes. And Leber congenital amaurosis, the main story in my gene therapy book , comes in at least 18 guises, LCA itself a subtype of retinitis pigmentosa.

A broken bone in a patient with OI type V. (Shakata GaNai)
Genetic heterogeneity has explained false accusations of child abuse. In osteogenesis imperfect (OI), aka “brittle bone disease,” bones break, often before birth. Like retinoblastoma, OI has left marks on history. An Egyptian mummy from 1000 B.C. had it, as did 9th century Viking “Ivan the Boneless,” who was reportedly comported into battle aboard a shield and whose remains were exhumed and burnt by King William I, forever obscuring the true diagnosis.
Until 1979, mutation in a single collagen gene was thought to cause OI. But cases arose of parents bringing babies with shattered bones to hospitals, and charges of child abuse filed. If social workers or other medical people knew about OI, the parents would be tested – but sometimes the tests were negative, even though the distraught parents insisted they hadn’t harmed their child.
Discovery of the second IO gene absolved some parents, but false accusations still happened. Then a third OI gene was found. I just checked Online Mendelian Inheritance in Man – the bible of geneticists – and found 14 types, but it’s confusing. Different types reflect mutations in the same gene, yet the same type can embrace different genes. The 14 types arise from 7 chromosomal addresses.
Whatever the terminology, the origins of discovering some of the recessive types are fascinating, often involving consanguinity (genetics speak for inbreeding).
OI type II was traced to the Mozobites, a polygamous sect in southern Algeria that had brother-sister/husband-wife pairs. Type VII came from a First Nations community in northern Quebec, where two generations of three interrelated families had many babies born with broken bones. Type IX families are from Pakistan, Senegal, and the Irish Traveller’s ethnic group and type XI derives from 5 consanguineous families from northern Turkey. Inbred families often illuminate rare single-gene diseases because parents share mutations inherited from their shared ancestors. The mutations are extremely rare in the outbred population in the area.
Annotation of the human genome is revealing more instances of genetic heterogeneity, explaining people who have clear symptoms of a known genetic disease yet test negative – like in retinoblastoma and osteogenesis imperfecta.
I hope the genetic testing industry, both clinical and direct-to-consumer, can keep up, and that patients/customers aren’t falsely reassured by low risks that don’t tell the whole story – because we may not know all of it, as was the case for retinoblastoma.
So while we’re busy sequencing all those exomes and genomes, keep in mind that we don’t know all there is to know. Single genes can still surprise us.
(For a more technical version of this information see Medscape.)


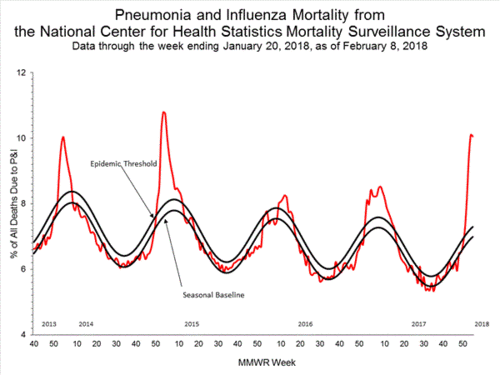 The weekly CDC flu report has been released, and it is full of important information. There were several things that jumped out at me, and I wanted to bring them to your attention.
The weekly CDC flu report has been released, and it is full of important information. There were several things that jumped out at me, and I wanted to bring them to your attention.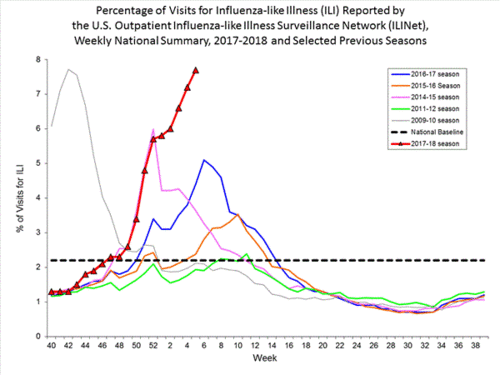 Second, and the CDC has just admitted this: This year's flu epidemic, in terms of hospital visits, has just surpassed 2009's H1N1 swine flu pandemic. Think of that! A non-pandemic strain of flu has sent more Americans to the hospital than the first pandemic in almost fifty years. While of course I am hopeful pediatric deaths will not reach the level seen in 2009, we are seeing pediatric and young adult sickness and death at a rate seldom seen in a flu season.
Second, and the CDC has just admitted this: This year's flu epidemic, in terms of hospital visits, has just surpassed 2009's H1N1 swine flu pandemic. Think of that! A non-pandemic strain of flu has sent more Americans to the hospital than the first pandemic in almost fifty years. While of course I am hopeful pediatric deaths will not reach the level seen in 2009, we are seeing pediatric and young adult sickness and death at a rate seldom seen in a flu season.














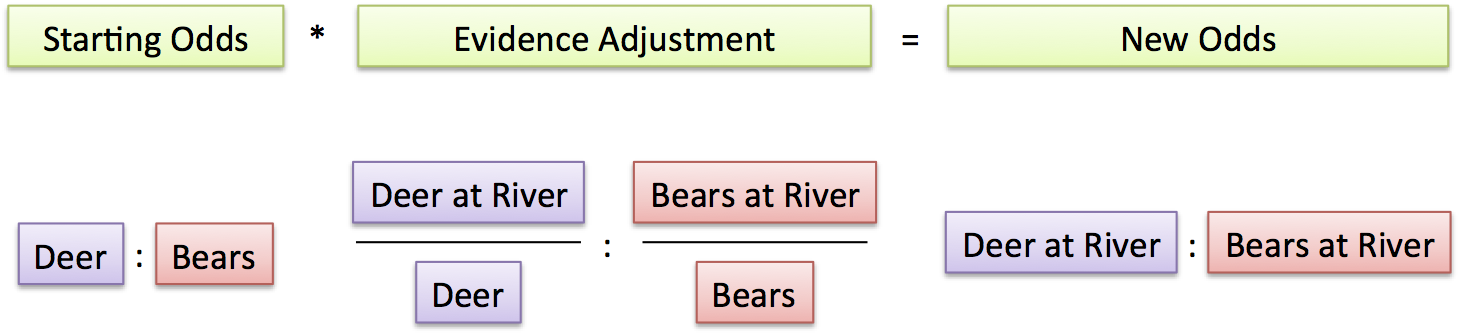
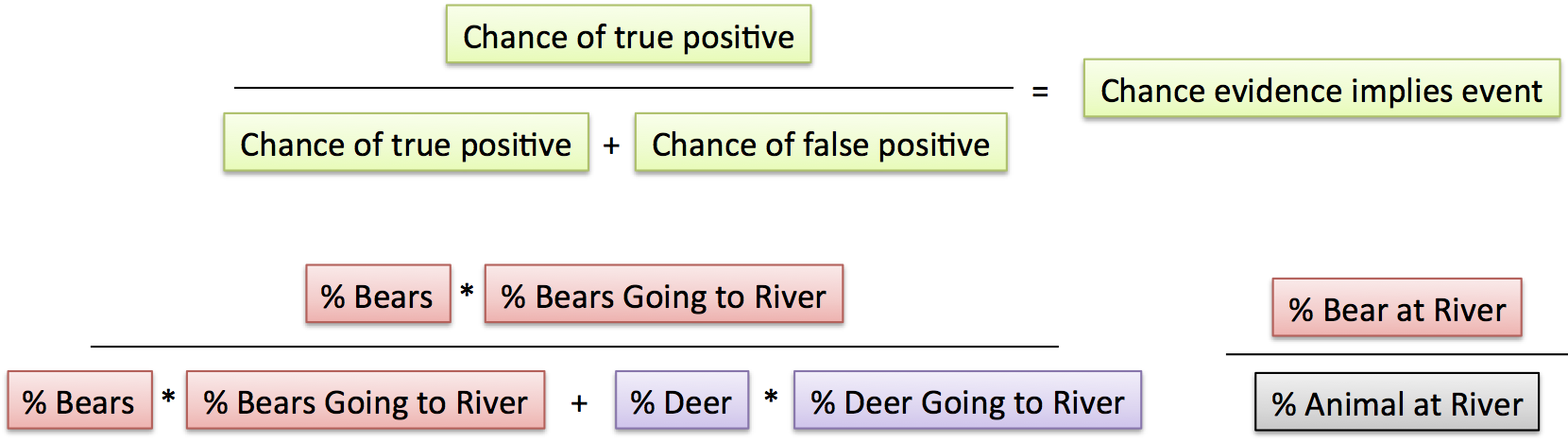
 = \frac{\Pr(\mathrm{X}|\mathrm{A})\Pr(\mathrm{A})}{\Pr(\mathrm{X|A})\Pr(A)+ \Pr(\mathrm{X|\sim A})\Pr(\sim A)}}}")