mjpdejong
Shared posts
The structure of DNA made visible
Genomics: Where the G-quadruplexes are
Nature Methods 12, 806 (2015). doi:10.1038/nmeth.3572
Author: Tal Nawy
A new sequencing method identifies G-quadruplexes genome wide.
Why a 5% Beer Gets You Drunk So Much Faster than a 4% Beer

If you look at the alcohol percentage on beers, it doesn’t seem like they’re all that different. After all, a 5% lager is just 1% more alcohol than a supposedly lighter beer, right? While that’s true, Draft Magazine explains the science of how ABV actually works.
Microsoft Data Harvesting Backported To Windows 7 & 8
Read the full post at darknet.org.uk
At 24, Linux Has Come Out of the Basement
 Happy Birthday, Linux Project -- this week you turned 24. The Linux OS has grown up everywhere. Its code and the open source model are found worldwide. People often use linux without knowing it -- when they search on Google, buy metro tickets or surf the Web. Linux powers all of that infrastructure. Linux travels worldwide on airplanes, and it's embedded in many of the smart devices that bring ultra convenience to our homes and cars. It runs our WiFi routers and our Android phones and tablets.
Happy Birthday, Linux Project -- this week you turned 24. The Linux OS has grown up everywhere. Its code and the open source model are found worldwide. People often use linux without knowing it -- when they search on Google, buy metro tickets or surf the Web. Linux powers all of that infrastructure. Linux travels worldwide on airplanes, and it's embedded in many of the smart devices that bring ultra convenience to our homes and cars. It runs our WiFi routers and our Android phones and tablets.
'Wetenschappelijke studies met korte titels krijgen meer aandacht'

Delftse wetenschappers verstrengelen deeltjes op 1,3 kilometer afstand

Strengths and limitations of your Nanodrop
Quantifying a DNA, RNA or protein sample concentration is now as easy as a click of the pipette, a push of a button and a dab of tissue to clean up. Here’s what you need to know about a few of the strengths and limitations of your Nanodrop – before you set up.
Take a number, please…
A Nanodrop is a common lab spectrophotometer (you may already be familiar with the 1000 or 2000 model) that reads a single 2μl drop on a pedestal. Less prep and cleanup time means you’re able to measure several samples in under a minute, compared to what’s needed to read just one sample in a traditional cuvette! This is perfect for most lab applications, but if you’re quantifying more than 96 samples at a time for microarrays, genotyping or to send off to a core facility, you might want to utilize a Nanodrop 8000 that can read up to 8 samples simultaneously.
Tell me the source: DNA, RNA or Protein
The Nanodrop does an excellent job at measuring across a wide spectrum that spans UV and visible light. It can’t automatically determine for you that the sample on the pedestal is DNA, RNA or protein – you have to tell the software before beginning measurements so it can report an accurate concentration. Though if you happen upon a circumstance where you’ve measured half of your RNA samples at the default setting (DNA for us) you can start over again by rereading each sample at the correct software setting, or you can estimate your sample concentration by hand, based upon the raw absorbance data already collected using the table below for guidance. Remember to correct for your blank TE or water sample!
The concentration at an A260 value of 1.0 for |
|
|---|---|
| Double stranded DNA | 50 μg/ml |
| Single stranded RNA | 40 μg/ml |
| Single stranded DNA | 33 μg/ml |
| Protein (measured at A280) | 1 mg/ml |
Please note that these are for relatively pure samples of DNA, RNA and protein at an A260/280 of about 1.8, 2.0 and 0.6, respectively. Additionally, the concentration of your sample will be reported in ng/μl.
Quick and dirty sample extractions: pump it up
Regardless of quantification method there are two important questions to ask after every nucleic acid extraction: Is there contamination in my sample tube? If there is, what kind is it?
From what may be read as a perfect DNA sample based upon the A260/280 alone, your Nanodrop may be overstating the results. This is a result common to most spectrophotometers: your equipment is doing what it was designed to do and that’s read absorbance at specific wavelengths.
But DNA is not the only item that absorbs at 260 nm. RNA does too. Phenol is close at about 270 nm and all of this combined has the potential to push your reported concentration upwards.
Become aware of contamination that you can see
Another point of interest is your A260/230 value, which like its A260/280 counterpart should land at or above 1.8 as a general rule of thumb, and above 2.0 for relatively pure DNA and RNA samples.
The Nanodrop paints a nice graphical picture to help you see just where attention may be needed
- Low A260/280, large peak at A280: protein contamination
- Large A230: phenol again, EDTA or carbohydrate contamination
- Small peaks or higher than expected absorbance along the far right side of the graph: contamination stemming from transfer of the interface during phase separation step in extraction.
- From the source, here’s what it can look like on your screen: 260/280 and 260/230 Ratios (ThermoScientific/Nanodrop).
One way to ensure a cleaner sample is to send it through to re-precipitation, followed by an ethanol wash, extended air-drying and re-suspension in a fresh volume of TE or pure water. Yes, your reported concentration may be significantly less than before, but that’s because you have successfully removed any contamination that was impacting your absorbance data for better or worse.
Look out below!
For the most part your nucleic acids extractions land within the measurable range of the Nanodrop (2ng – 15μg per μl). It’s when you start extracting from single cell or small sample sources that you need to consider alternative quantification methods. For example, fluorescent-dyes will help resolve amounts of DNA or RNA at or below 2ng/μl.
Want to learn more about DNA, RNA and your Nanodrop? Check out these other interesting resources:
- Nucleic Acid Quantitation (Wikipedia)
- Nanodrop (OpenWetWare)
- Quick reference: Determining DNA Concentration & Purity
- Spectrophotometer for nucleic acid measurements
- Assessment of Nucleic Acid Purity
- Where did my DNA go? Tips on improving DNA Clean-Up
- DNA Precipitation: Ethanol vs. Isopropanol
- Ethanol Precipitation of DNA and RNA: How it works
Another Evil TV Geneticist on Netflix’s “Between”

 Ten years ago this month, I attended the Catalyst Workshop at the American Film Institute. The week-long program taught screenwriting to a dozen scientists, with the hope that we’d somehow help Hollywood get the science right.
Ten years ago this month, I attended the Catalyst Workshop at the American Film Institute. The week-long program taught screenwriting to a dozen scientists, with the hope that we’d somehow help Hollywood get the science right.
But what we learned during that fabulous week, dissecting films such as “The Day After Tomorrow,” meeting top studio people and even pitching ideas, is that the science just doesn’t matter. Although I agree that story and characters are paramount, getting the science right doesn’t take much effort, and can not only improve plausibility, but also accurately portray scientists. Geneticists in particular seem especially vulnerable to villianizing.
A TIRED PLOT
My latest target for tarnishing the image of geneticists is “Between,” a six-episode Netflix offering from Canada. (Yes, I know Halle Berry’s hybrid son on Extant has something to do with a virus, but I gave up two episodes ago.)
“Between” is an amalgam of familiar themes. It’s Logan’s Run, Under The Dome, and Lord of the Flies, with a small town setting like that of Wayward Pines, which I blogged about recently. And like those shows, the focus is on the characters and their relationships, with the predicament a backdrop that fuels the interactions.
So now we have Between. In the town of Pretty Lake, over the course of a few days, everyone aged 22 and older suddenly gasps, bleeds from the nose, and dies. The government quarantines the town and the young folk pile up the bodies of their elders and have a huge bonfire. Early in episode one, a main character, a geeky young man named, of course, Adam, mentions his father, a mysteriously missing geneticist. This is repeated throughout the early episodes. Foreshadow the mad scientist.
A nice touch is the accurate depiction of a major character who has Down syndrome. But one scene dangerously got insulin shock ass backwards. A boy with type 1 diabetes collapses from days without insulin and everyone yells “He’s in insulin shock! Give him candy!” Without insulin, how would his cells take up the glucose? Who vetted that script?
 BLAME THE VIRUS
BLAME THE VIRUS
Soon, the people in Pretty Lake and the talking heads on their screens deem the culprit a virus. My husband Larry (a chemist) and I hung on through all six episodes to learn exactly how a virus could tell that a person has reached age 22.
We learn in the final episode, when the crazy nameless geneticist shows up via a tunnel that perhaps comes from Wayward Pines, that he engineered the virus. He cleverly included a bit of his own DNA so that he and his son would be immune. Note to the show’s writers: a parent and a child are not genetically identical. Nor do they have the same exposures guiding development of their immune systems.
The evil geneticist was working alone on the virus, because the government sends in unwitting workers wielding needles to “vaccinate” the town’s young survivors, supposedly so the quarantine can be lifted. But no. The shots will kill the young people to protect the rest of the world, as the clueless shotgivers over age 21 become victims, bleeding and dropping.
Larry and I were sorely disappointed with the ending, which did nothing to satisfy the craving of the scientifically-minded to understand how things work.
The geneticist explains how the virus kills people over 21 with a lot of posturing, emitting a barrage of inappropriate technical terms, concluding inexplicably with something about the ozone layer. It all speeds by too fast to process, but the spouting renegade geneticist seems to have invented the virus to fight overpopulation. I think. The focus is much more on teen angst involving a pregnancy and jealousy than how a virus can tell how old someone is.
When Adam parrots his dad’s message to the others, that the killer virus homes in on “the biological clock,” everyone nods with comprehension. But that term refers to circadian and other biorhythms, not a FitBit like contraption embedded in our spleens that flashes our exact ages. Basic Bio 101 writers! But it’s easy to see how that sort of error might have come up: oversimplification in a news release, which means oversimplification in the articles that spawn from it. The news release headline “Leicester scientists to unlock the secrets of the biological clock” actually refers to telomeres, the tips of chromosomes that whittle down with increasing age. Aha!

TELOMERES ANYONE?
The only reason that I watched “Between” to begin with was in anticipation of using telomeres in an apocalypse story. Shrinking chromosome tips are like cellular tree rings in reverse, disappearing rather than accruing with the passage of time. With repeats of TTAGGG lopped off one by one from chromosome ends as time goes on, marking the number of cell divisions, telomeres are more quantitative than gray hairs, achy joints, and wrinkles.
In telomeres, Nature provided the perfect plot point, if only the show’s writers had looked.
The telomere clock dates back to the famous “Hayflick Limit” of 40 to 60 mitoses for cells in culture. In the 1950s, Leonard Hayflick, PhD, was a young researcher at the Wistar Institute in Philadelphia. He wanted to see what the fluid around cancer cells would do to normal cells from human embryos and fetuses. At that time all cells were thought to be immortal, dividing unceasingly. “Today if I did that using federal research dollars to grow tissue from human embryos or fetuses, I would go to jail,” he told me a few years ago for my essay collection.
 Hayflick had wanted to run replicates of his experiment simultaneously, but the supply of material was erratic. “In my incubator at any given time, I’d have 12 to 20 cultures going, each marked with a different start date.”
Hayflick had wanted to run replicates of his experiment simultaneously, but the supply of material was erratic. “In my incubator at any given time, I’d have 12 to 20 cultures going, each marked with a different start date.”
He soon discovered something strange. “Despite the fact that I used the same technician, the same glassware, and the same media, the cells in culture the longest stopped dividing, while the young cultures luxuriated. That shouldn’t happen. It intrigued me, so I began to look at what was going on.”
Hayflick’s fetal cells died after being moved a certain number of times, each move triggering cell division. He and colleague Paul Moorhead repeated their astonishing experiments over and over, with the same results – cells obeyed some sort of internal clock that marks the number of cell divisions. Freeze cells at division 20, and when thawed, they’d pick up where they left off, dividing 20 to 40 more times.
But it’s hard to change dogma and get published. And so Hayflick and Moorhead “sent the luxuriating cultures – the young ones – to the grey eminences of the field. We would tell them, ‘by May 20th to 30th, the cells are going to die.’ When the phone started ringing between May 20th and 30th, we decided to publish. If our work went up in flames, we’d be in the company of the grey eminences,” Hayflick told me. After a stinging rejection from one journal, the findings were published in Experimental Cell Research in 1961.
Discovery of the Hayflick limit founded what we now call telomere biology. Many others, including Elizabeth Blackburn, Carol Greider, and Jack Szostack, who shared the Nobel Prize for Physiology or Medicine in 2009, discovered the DNA sequence of human telomeres and how they function as fuses, marking biological time. DNA polymerase cannot replicate DNA at the end of a strand, unless an enzyme called telomerase tacks on more repeats – as happens in cancer cells. So Pretty Lake might have had a few cancer patients hanging around with the quarreling teens and kids. Their chromosome tips would appear young.
MECHANISM?

Larry and I eagerly awaited a character turning 22 to find out how the virus tracks time. Indeed, a pretty and popular teacher celebrates her 22nd birthday midway through episode 4, and sure enough, within minutes, spews nasal blood and expires. How could that have happened? After all, telomere shrinkage is not a discrete and uniform phenomenon. All 21-year-olds don’t have x number of TTAGGGs and a 22-year-old x-1.
I’m imagining deploying a virus that targets the telomeres using a form of genome editing (CRISPR/cas-9, TALENs, or zinc finger nucleases) and only integrating into a host chromosome if the number of repeats is below a certain threshhold. Then, the virus induces hemorrhage, perhaps by turning off transcription of various clotting factors. (Readers please elaborate or pose alternate hypotheses.)
Reviewers trashed Between for a lot of reasons – too derivative, “ho-hum,” a “familiar ensemble soap opera with conspiracy-theory embroidery,” and Hollywood Reporter’s “It’s the end of the world as they know it, and viewers won’t care.” None that I could find mentioned the spotty science.

I’m disturbed about the missed opportunity to imagine how a virus could take out an entire huge age cohort of humanity. But I’m much more disturbed about the tired stereotype of the mad scientist, especially the ego-driven geneticist who tailors a mysterious and dangerous virus to control human population growth. It’s not only absurd, but in this age of Ebola epidemics, and fear of vaccines and genetic modification, the ideas behind Between and the events at Pretty Lake are downright dangerous.
Desktop Best Buy Guide - September 2015
Learn-omics! What is that “Omics” I keep Stumbling Upon?
Genomics, transcriptomics, proteomics, metabolomics – words that in 2015 sound very familiar even to a freshman in any biology field. Although most have heard those words before, I keep encountering students or even post-graduates who find it difficult to explain what they are. So, to make things easier here is a peek behind the curtains of “omics technologies”.
Word origin
In order for someone to be able to explain something, he or she should know where it comes from.
As we all know, scientists keep using Latin and Greek terminology in various fields and this also applies to our predicament.
Even from ancient Greece, medical terms used the suffix –ome, which later passed to Latin as variant –oma (i.e. lipoma, melanoma) and when in biology the word gene emerged, researchers quickly used the word genome to easily encapsulate a field of studies in a single word.
So, from a linguistic point of view, genome came first, and genomics after. Genomics is used to emphasize the technologies which were used to study the…genome, and if you are wondering which genome, well that’s yours.
Yes! With the human genome project being all the rage in the 90s, the term genomics was used for the first time by the consortium that was assembled in 1986. More specifically, after a long day mapping part of the human genome, beer and McDonald’s was the magic combination for Tom Roderick to propose the word to his colleagues, Frank Ruddle and Victor McKusick. He proposed it only as an idea to call a potential new scientific journal of theirs, which today is well known as Genomics.
Genomics was the first “omics” terminology used, far earlier than the second in 1995, proteomics, and so on.
So there you are, now you know where “Omics” come from, but why are they so important?
The need of inventing “Omics”
The need to create these words rose from the need to study holistic (that’s right people, another Greek word) or, if you prefer, the totality of the biology of the systems (systems biology). Soon, scientists understood that studying one gene or one metabolite or one mRNA gives little knowledge. Modern technology allows scientists to study the whole transcriptome, genome or proteome of an organism giving more and more information, so now the challenge is not to generate data, but to interpret them and to relate results of different omics technologies (i.e. trancriptomics with metabolomics).
As the technology advances and new ideas come forth, new technologies and terminologies arise. Phenomics, lipidomics, catalomics, connectomics are some of them, translating to the holistic approach of studying phenotypes, lipids, enzymes and neural connections.
So, stop thinking gene by gene, enzyme by enzyme, metabolite by metabolite and broaden your horizons to apply omics technologies for a wider knowledge of systems biology. Here are some examples on how to achieve it.
Genomics
The technologies that are focused on studying the genome of an organism including genes, exons, introns, promoters, transcription factors, and many other genome-related functions.
Technologies used: Next generation sequencing (i.e. Illumina HiSeq), Sanger sequencing, de novo assembling, bioinformatics.
Transcriptomics
The technologies that are focused on studying the transcriptome of an organism.
Technologies used: Next generation sequencing (RNA sequencing), bioinformatics, microarrays, real-time PCR.
Proteomics
The technologies that are focused on studying the proteins of an organism or the proteome.
Technologies used: Enzyme-linked immunosorbent assay (ELISA), mass spectrometric immunoassay (MSIA)
Metabolomics
The technologies that are focused on studying the metabolome or the secondary metabolites of an organism, such as amino acids, sugars, phosphates, nitrogen containing compounds, polyols, etc.
Technologies used: Gas chromatography fused with mass spectrometry (GC-MS), liquid chromatography fused with mass spectrometry (LC-MS).
Lipidomics
The technologies that are focused on studying the cellular lipids of an organism or a biological system.
Technologies used: Electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI).
Catalomics
The technologies that are focused on studying enzymes (catalysts) or other biocatalysts of an organism or a system.
Technologies used: “Click” reactions in chemistry.
I think you might have stopped reading by now, so these are only some examples of “Omics” technologies and ways to approach them. This is just a brief glimpse into the technology that is here to stay. Now, it is in our hands to exploit it so basic and applied research can reap the benefits.
“Before beginning a Hunt, it is wise to ask someone what you are looking for before you begin looking for it.”
Winnie the Pooh
References
- Babiniotis G. (2009). The history of the words.
- Daniel C. Liebler (2002). Introduction to proteomics: tools for the new biology. Totowa, NJ: Humana Press.
- Karunakaran A. Kalesh et. al.(2010). The use of click chemistry in the emerging field of catalomics. Org Biomol Chem. 8(8):1749-62
- Kopka J. (2006). Current challenges and developments in GC-MS based metabolite profiling technology. J Biotechnol. Jun 25;124(1):312-22.
- McGettigan PA. (2013). Transcriptomics in the RNA-seq era. Curr Opin Chem Biol. Feb;17(1):4-11.
- Yadav SP. (2007). The Wholeness in Suffix -omics, -omes, and the Word Om. Journal of Biomolecular Techniques. Dec 18(5):277.
- McCouch SR. (2001). Genomics and Synteny. Plant. Physio. Jan;125(1):152-5.
- Wenk MR (2005). The emerging field of lipidomics. Nat Rev Drug Discov 4 (7): 594–610.
Nicotine changes marijuana’s effect on the brain
Filbey, F., McQueeny, T., Kadamangudi, S., Bice, C., & Ketcherside, A. (2015) Combined effects of marijuana and nicotine on memory performance and hippocampal volume. Behavioural Brain Research, 46-53. DOI: 10.1016/j.bbr.2015.07.029
Combined effects of marijuana and nicotine on memory performance and hippocampal volumeBiochemici slaan data lange tijd op in DNA
Will Microsoft block Windows 10 users from playing counterfeit games?
New Insights into Human De Novo Mutations
Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I, Genome of the Netherlands Consortium, van Duijn CM, Swertz M, Wijmenga C.... (2015) Genome-wide patterns and properties of de novo mutations in humans. Nature genetics, 47(7), 822-6. PMID: 25985141
Genome-wide patterns and properties of de novo mutations in humans.The ones that get away—does intensive fishing make fish harder to catch?
Shaun S. Killen, Julie J. H. Nati, & Cory D. Suski. (2015) Vulnerability of individual fish to capture by trawling is influenced by capacity for anaerobic metabolism. Proceedings of the Royal Society B. info:/10.1098/rspb.2015.0603
Intels Skylake-platform: sneller maar niet zuiniger
How DNA Extraction Kits Work in the Lab
We give a lot of troubleshooting help on RNA and DNA extraction here at Bitesize Bio because almost everything we do in molecular biology requires DNA or RNA at the very first step. These days, most labs use commercial DNA extraction kits, which employ spin columns, for the isolation of DNA and RNA. The spin columns contain a silica resin that selectively binds DNA and RNA, depending on the salt conditions and other factors influenced by the extraction method.
These DNA extraction kits make the whole process much easier and faster than the methods of old, when things are going well, but the downside of using a kit is that if you don’t understand what is in the black box of the kit, it makes troubleshooting much more difficult.
So in this article, I’ll explain in some detail how RNA and DNA extraction kits work and what is going on at each step. I’ll also go over some common problems specific to using silica columns in DNA extraction that can be overcome or avoided with just a little extra understanding.
The First Step in RNA and DNA Extraction: Lysis
The lysis formulas may vary based on the whether you want to extract DNA or RNA, but the common denominator is a lysis buffer containing a high concentration of chaotropic salt. Chaotropes destabilize hydrogen bonds, van der Waals forces, and hydrophobic interactions. Proteins are destabilized, including nucleases, and the association of nucleic acids with water is disrupted setting up the conditions for the transfer to silica.
Chaotropic salts include guanidine HCL, guanidine thiocyanate, urea, and lithium perchlorate.
Besides the chaotropes, there is usually some detergents in the lysis buffer to help with protein solubilization and lysis. There can also be enzymes used for lysis depending on the samples type. Proteinase K is one of these, and actually works very well in these denaturing buffers; the more denatured the protein, the better Proteinase K works. Lysozyme, however, does not work in the denaturing and so lysozyme treatment is usually done before adding the denaturing salts.
One comment about plasmid preps, the lysis is very different than extraction for RNA or genomic DNA extraction because the plasmid has to be separated from the genomic DNA first and if you throw in chaotropes, you’ll release everything at once and won’t be able to differentially separate the small circular DNA from the high molecular weight chromosome. So, in plasmid preps the chaotropes are not added until after lysis and the salts are used for binding. An excellent in-depth article on alkaline lysis is here and also another article on the difference between genomic DNA and plasmid is available for further reading.
After DNA Extraction Comes Purification: Binding the DNA to the Column
The chaotropic salts are critical for lysis, but also for binding the DNA (or RNA) to the column, as we discussed. Additionally, to enhance and influence the binding of nucleic acids to silica, alcohol is also added.
Most of the time this is ethanol but sometimes it may be isopropanol. The percent ethanol and the volume has big effects. Too much and you’ll bring in a lot of degraded nucleic acids and small species that will influence UV260 readings and throw off some of your yields. Too little, and it may become difficult to wash away all of the salt from the membrane.
The important point here is that the ethanol influences binding and the amount added is optimized for whatever kit you are using. Modifying that step can help change what you recover so if you are having problems and want to troubleshoot recovery, that can be a step to evaluate further.
Another way to diagnose problems is to save the flow-through after binding and precipitate it to see if you can find the nucleic acids you are searching for. If you used an SDS-containing detergent in lysis, try using NaCl as a precipitant to avoid contamination of the DNA or RNA with detergent.
Washing the DNA (or RNA)
Your lysate was centrifuged through the silica membrane and now your extracted DNA or RNA should be bound to the column and the impurities, protein and polysaccharides, should have passed through. But, the membrane is still dirty with residual proteins and salt. If the sample was from plants, there will still be polysaccharides, maybe some pigments too, left on the membrane, or if the sample was blood, the membrane might be tinted brown or yellow.
The wash steps serve to remove these impurities. There are typically two washes, although this can vary depending on the sample type. The first wash will often have a low amount of chaotropic salt to remove the protein and colored contaminants. This is always followed with an ethanol wash to remove the salts. If the prep is something that didn’t have a lot of protein to start, such as plasmid preps or PCR clean up, then only an ethanol wash is needed.
Removal of the chaotropic salts is crucial to getting high yields and purity DNA or RNA. Some kits will even wash the column with ethanol twice. If salt remains behind, the elution of nucleic acid is going to be poor, and the A230 reading will be high, resulting in low 260/230 ratios.
For Ethanol-Free DNA and RNA, You Need a Dry Spin
After the ethanol wash, most protocols have a centrifugation step to dry the column. This is to remove the ethanol and is essential for a clean eluant. When 10 mM Tris buffer or water is applied to the membrane for elution, the nucleic acids can become hydrated and will release from the membrane. If the column still has ethanol on it, then the nucleic acids cannot be fully rehydrated.
Skipping the drying step results in ethanol contamination and low yields. I do not see ethanol absorbance on the Nanodrop, so it won’t show up in your readings. The main indicators of a problem are that when you try to load the sample onto an agarose gel, the DNA will not sink. Even in the presence of loading dye. Another indicator is that if you put the sample in the -20C, it doesn’t freeze.
RNA and DNA Extraction, the Final Frontier: Elution
The final step in the DNA extraction protocol is the release of pure DNA or RNA from the silica.
For DNA preps, 10 mM Tris at a pH between 8-9 is typically used. DNA is more stable at a slightly basic pH and will dissolve faster in a buffer. This is true even for DNA pellets. Water tends to have a low pH, as low as 4-5 and high molecular weight DNA may not completely rehydrate in the short time used for elution. Elution of DNA can be maximized by allowing the buffer to sit in the membrane for a few minutes before centrifugation.
RNA, on the other hand, is fine at a slightly acidic pH and so water is the preferred diluent. RNA dissolves readily in water.
What Other Things Can Go Wrong with RNA and DNA Extraction
Low yields: If you experience DNA / RNA yields lower than you expected for a sample, there are many factors to think about. Usually it is a lysis problem. Incomplete lysis is a major cause of low yields. It could also be caused by incorrect binding conditions. Make sure to use fresh high quality ethanol (100% 200 proof) to dilute buffers or for adding to the binding step. Low quality ethanol or old stocks may have taken on water and not be the correct concentration. If the wash buffer is not made correctly, you may be washing off your extracted DNA or RNA.
Low Purity: If the extracted DNA is contaminated with protein (low 260/280) then maybe you started with too much sample and the protein was not completely removed or dissolved. If the DNA has poor 260/230 ratio the issue is usually salt from the bind or the wash buffer. Make sure that the highest quality ethanol was used to prepare wash buffers and if the problem continues, give the column an additional wash.
Some samples have a lot more inhibitors compared to others. Environmental samples are especially prone to purity issues because humic substances are solubilized during extraction. Humics behave similarly to DNA and are difficult to remove from the silica column. For this type of sample, specialized techniques exist to remove the protein and humics prior to the column step.
Degradation: This is more of a concern for RNA preps and an article that gives specific advice is here. Mainly with RNA extractions, degradation occurs from improper storage of the sample or an inefficient lysis, assuming of course that you eluted with RNase-free water. For DNA extractions, degradation is not a huge problem because for PCR, the DNA can be sheared and it works fine. But if you were hoping to not have so much sheared DNA, then you may have used too strong a lysis method.
PCR Clean-up Special Considerations: PCR cleanup obviously isn’t a DNA extraction technique per se, but it is a nice and easy techniques, because it is simply adding a high concentration of binding salts (typically between 3-5 volumes of salt per volumes of PCR reaction) and centrifugation through the column. So when PCR Clean-up kits fail, it can be particularly frustrating. The first question I ask people is “did you check the results of the PCR on a gel?” because you cannot UV check a PCR reaction and have an accurate reading. There is way too much in a PCR reaction absorbing UV at 260: nucleotides, detergents, salts, and primers. In my experience, a failure of a PCR clean-up kit to work frequently is caused by a PCR reaction that has failed and so there was nothing to clean up. But if you know you had a strong PCR product, the best approach is to just save your flow-through fraction after binding. If the DNA doesn’t bind, that’s where it is. You can always rescue it and then clean it up again. And then call tech support and ask for a replacement kit.
Go Forth and Perform Your RNA and DNA Extractions with Confidence
As scientists, of course we want to know exactly what is going on with our experiments and be able to troubleshoot without having to call technical service first.
I hope that this article helps clarify some of the science around the silica spin filter method for RNA and DNA extractions so you can make your own diagnosis and fixes. So, when you do call technical service, you’ll have double checked a few of the most likely causes of problems first and instead of going through a lot of rigmarole, you can get to a resolution much faster. Even if that is a free replacement DNA extraction kit!
Any questions? Any other problems with silica spin filter preps that you don’t understand? Let us know or ask a question in our “Questions” section and we’ll discuss!
Originally published on June 28, 2010. Updated and revised on July 11, 2015.
Rare form: Novel structures built from DNA emerge
Modified DNA building blocks are cancer's Achilles heel
Linux still rules supercomputing
How to Become Immortal: Generation of Immortal Cell Lines
Normal cells are unable to replicate past several rounds of proliferation (termed the Hayflick limit) as with each round of proliferation the telomeres shorten. When the telomeres reach a critically reduced length, DNA damage is triggered leading to cellular senescence.
Therefore, if you tried to culture a primary cell population it would eventually die unless the cells were manipulated in some way to circumvent the process of senescence and become immortal. Here we discuss various ways to overcome the hayflick limit and induce immortality in cultured cells.
Sources of Imortality
Spontaneously Immortalized Cells
The best example of this would be cancer cells, which may have undergone genetic changes to resist senescence and are immortal. However, many cancer cell lines may not have these changes, in fact, George Gey, the scientist who created the first immortalized and arguably the most famous cell line: HeLa cells, had to test hundreds of cancer lines before stumbling upon the highly metaplastic ovarian cells of Henrietta Lacks. Thus, other methodologies may be required to help even cancer cells become immortal.
Introduce a Viral Gene that Overrides the Cell Cycle
Many viral genes affect the cell cycle and thus can be used to overcome senescence by removing the biological brakes on proliferative control. Many of these are tumor suppressor genes, since they require suppression for tumorigenesis to occur. The most common of these is over-expression of the Large T-antigen of the simian virus (SV-40), which represses the retinoblastoma (Rb) and p53 genes, both critical controllers of the cell cycle.
One example of a cell line immortalized with SV40 is HEK293T, which are also known as 293T cells, a cell line widely used to express viral particles and for many cellular assays, since they are very easily transfected. Other viral genes include those from the human papilloma virus (HPV) such as E6 and E7, which also target Rb and p53.
Expression of Genes that Confer Immortality
The most well-known immortality gene is Telomerase (hTERT). A ribonucleoprotein, telomerase is able to extend the DNA sequence of telomeres, thus abating the senescence process and enabling the cells to undergo infinite cell divisions. Indeed, telomerase has recently been heralded as a potential mechanism to reverse aging. The issue with using telomerase to reverse ageing is that increased telomerase expression can also induce tumor growth. Indeed, many of the same genes used to create immortalized cell lines such as hTERT and SV40 induce tumor formation. Thus, it is unsurprising that hTERT is often found over-expressed in human tumors, thus imparting one of the key hallmarks of cancer: unrestrained proliferation. Exactly what we use it for when immortalizing a primary cell line!
Combining Tumor Suppressor Inactivation and Telomerase Expression
In some cell types, using only one immortalization method may yield low numbers of cells that have become immortal. Therefore, depending on your cell line, it may be beneficial to combine both suppression of a tumor suppressor (such as the cell cycle inhibitors mentioned above) and expression of hTERT to immortalize a larger number of cells. This dual method is also suggested if you wish to both immortalize and transform (i.e. make tumorigenic-like) your cell line, as evidence suggests that some cell lines do not undergo efficient transformation without some immortalization.
How to Introduce Immortality to a Primary Cell Line
Many of the above sources of immortality are based on genetic manipulation of your primary cell line. This requires the introduction of foreign DNA into your cells. As many primary cells lines are frustratingly difficult to transfect, the easiest and most effective way to introduce genetic changes into a primary cell line is through viral infection.
The most popular method is through replication-deficient lentiviruses, since they are relatively safer then adenoviruses which have the ability to re-infect cells and thus contain live virus for a much longer period of time. If you want more information on lentiviral production, check out the Bitesize Bio Webinar and the Addgene Lentiviral Resource Page. Retroviruses can also be used to transfect cells, however, they can only infect actively dividing cells, thus reducing the number of cells that may be transduced with virus.
Words of Caution: Immortality May Not be the Best Route!
By introducing genetic changes into your cells, you may be profoundly altering the phenotype of your cell line. Although this does make your cell line more useful in some ways: it may make your cells more homogeneous allowing for replication of results, you can create large stocks of cells for future use and they may be easier to experimentally, there is still many benefits to using primary cell lines.
As primary lines adapt to being culture and being immortalized, cell populations and cellular mechanisms are altered. This may confound your experiments and lead to inconclusive or erroneous results. In addition, there is much debate about how accurately immortalized cells model real tissue. For example, although controversial, it is unlikely that SV40 can infect humans, although the mechanism of action of SV40: p53 and Rb mutations can be very common mutations in human tumors. Thus, it is important to identify the best genetic manipulation to use to immortalize your cell type.
Therefore, although immortalized cells are immensely powerful in their utility for experimental research, some caveats must be accepted in their use. As with any experimental model, immortalized cells are simply a model for your intended cellular system.
Additional Reading:
Rebecca Skloot – “The Immortal Life of Henrietta Lacks”
This is a must-read for all budding and experienced biologists. A well-researched book describes the identification of HeLa cells, their scientific value as well as the ethical dilemmas they have identified and the stress placed on the Lacks family.
References:
ATCC: HEK293T cells (ATCC CRL-3216)
Garbe et al (1999) Viral oncogenes accelerate conversion to immortality of cultured conditionally immortal human mammary epithelial cells. Oncogene. 18(13): 2169
Callaway, E (2010) Nature News: Telomerase reverses ageing process.
Hanahan & Weinberg (2011) Hallmarks of Cancer: The Next Generation. Cell. 144(5): 646.
Zhu et al (1991) The ability of simian virus 40 large T antigen to immortalize primary mouse embryo fibroblasts cosegregates with its ability to bind to p53. Journal of Virology. 65(12): 6872.
Redell (1999) The role of senescence and immortalization in carcinogenesis. Carcinogenesis. 21(3): 477.
BitesizeBio Webinar: Lentiviruses 101: Plasmids and Viral Production.
Addgene Lentiviral Protocols and Resources
Nelson (2001) Debate on the Link Between SV40 and Human Cancer Continues. Journal of the National Cancer Institute. 93(17): 1284
Sherr and McCormick (2002) The RB and p53 pathways in cancer. Cancer Cell. 2(1): 103.
CloudFlare Fights Cancer, Or the Unexpected Benefits of Open Sourcing Your Code
|
Sjon
shared this story
from |
Recently I was contacted by Dr. Igor Kozin from The Institute of Cancer Research in London. He asked about the optimal way to compile CloudFlare's open source fork of zlib. It turns out that zlib is widely used to compress the SAM/BAM files that are used for DNA sequencing. And it turns out our zlib fork is the best open source solution for that file format.
 CC BY-SA 2.0 image by Shaury Nash
CC BY-SA 2.0 image by Shaury Nash
The files used for this kind of research reach hundreds of gigabytes and every time they are compressed and decompressed with our library many important seconds are saved, bringing the cure for cancer that much closer. At least that what I am going to tell myself when I go to bed.
This made me realize that the benefits of open source go much farther than one can imagine, and you never know where a piece of code may end up. Open sourcing makes sophisticated algorithms and software accessible to individuals and organizations that would not have the resources to develop them on their own, or the money pay for a proprietary solution.
It also made me wonder exactly what we did to zlib that makes it stand out from other zlib forks.
Recap
Zlib is a compression library that supports two formats: deflate and gzip. Both formats use the same algorithm also called DEFLATE, but with different headers and checksum functions. The deflate algorithm is described here.
Both formats are supported by the absolute majority of web browsers, and we at CloudFlare compress all text content on the fly using the gzip format. Moreover DEFLATE is also used by the PNG file format, and our fork of zlib also accelerates our image optimization engine Polish. You can find the optimized fork of pngcrush here.
Given the amount of traffic we must handle, compression optimization really makes sense for us. Therefore we included several improvements over the default implementation.
First of all it is important to understand the current state of zlib. It is a very old library, one of the oldest that is still used as is to this day. It is so old it was written in K&R C. It is so old USB was not invented yet. It is so old that DOS was still a thing. It is so old (insert your favorite so old joke here). More precisely it dates back to 1995. Back to the days 16-bit computers with 64KB addressable space were still in use.
Still it represents one of the best pieces of code ever written, and even modernizing it gives only modest performance boost. Which shows the great skill of its authors and the long way compilers have come since 1995.
Below is a list of some of the improvements in our fork of zlib. This work was done by me, my colleague Shuxin Yang, and also includes improvements from other sources.
-
uint64_tas the standard type - the default fork used 16-bit types. - Using an improved hash function - we use the iSCSI CRC32 function as the hash function in our zlib. This specific function is implemented as a hardware instruction on Intel processors. It has very fast performance and better collision properties.
- Search for matches of at least 4 bytes, instead the 3 bytes the format suggests. This leads to fewer hash collisions, and less effort wasted on insignificant matches. It also improves the compression rate a little bit for the majority of cases (but not all).
- Using SIMD instructions for window rolling.
- Using the hardware carry-less multiplication instruction
PLCMULQDQfor the CRC32 checksum. - Optimized longest-match function. This is the most performance demanding function in the library. It is responsible for finding the (length, distance) matches in the current window.
In addition, we have an experimental branch that implements an improved version of the linked list used in zlib. It has much better performance for compression levels 6 to 9, while retaining the same compression ratio. You can find the experimental branch here.
Benchmarking
You can find independent benchmarks of our library here and here. In addition, I performed some in-house benchmarking, and put the results here for your convenience.
All the benchmarks were performed on an i5-4278U CPU. The compression was performed from and to a ramdisk. All libraries were compiled with gcc version 4.8.4 with the compilation flags: "-O3 -march=native".
I tested the performance of the master zlib fork, optimized implementation by Intel, our own master branch, and our experimental branch.
Four data sets were used for the benchmarks. The Calgary corpus, the Canterbury corpus, the Large Canterbury corpus and the Silesia corpus.
Calgary corpus
Performance:
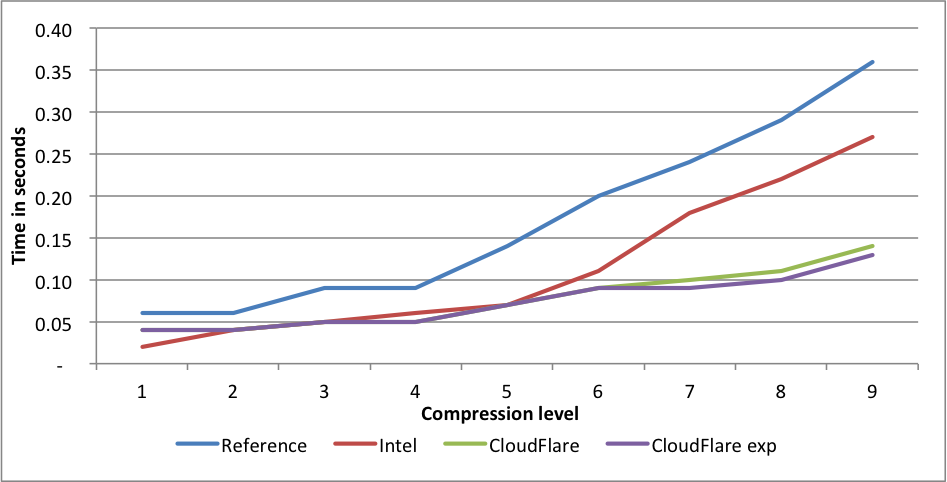 Compression rates:
Compression rates:
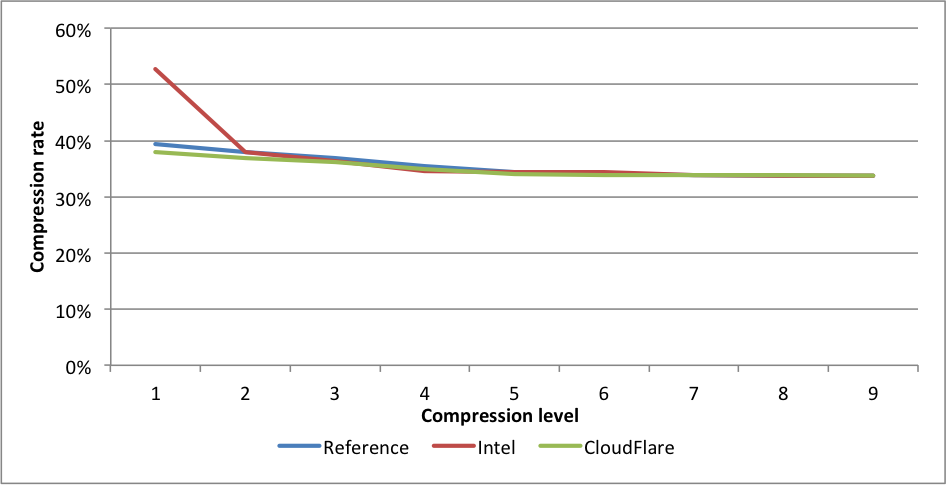 For this benchmark, Intel only outperforms our implementation for level 1, but at the cost of 1.39X larger files. This difference is far greater than even the difference between levels 1 and 9, and should probably be regarded as a different compression level. CloudFlare is faster on all other levels, and outperforms significantly for levels 6 to 9. The experimental implementation is even faster for those levels.
For this benchmark, Intel only outperforms our implementation for level 1, but at the cost of 1.39X larger files. This difference is far greater than even the difference between levels 1 and 9, and should probably be regarded as a different compression level. CloudFlare is faster on all other levels, and outperforms significantly for levels 6 to 9. The experimental implementation is even faster for those levels.
Canterbury corpus
Performance:
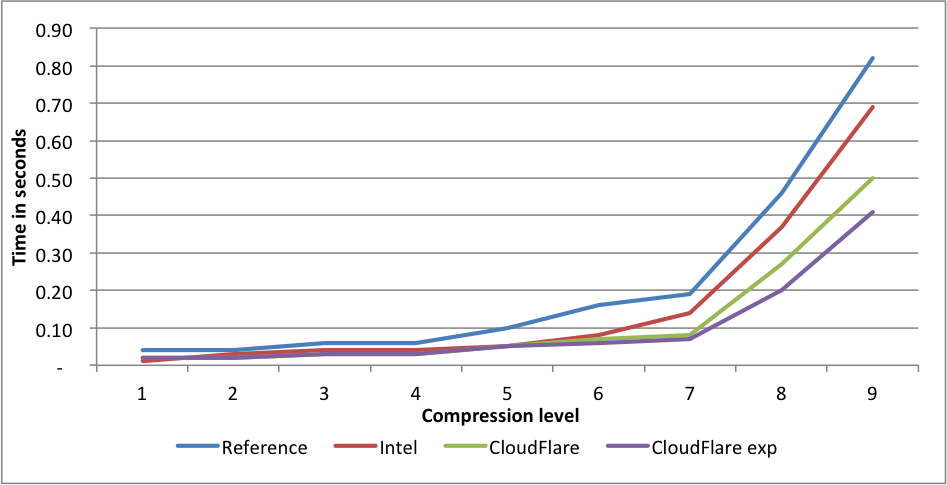 Compression rates:
Compression rates:
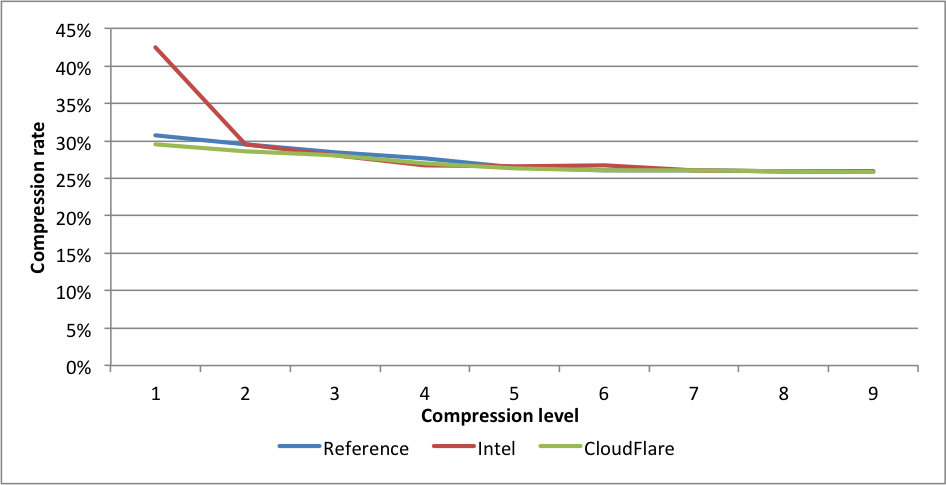 Here we see a similar situation. Intel at level 1 gets 1.44X larger files. CloudFlare is faster for levels 2 to 9. On level 9, the experimental branch outperforms the reference implementation by 2X.
Here we see a similar situation. Intel at level 1 gets 1.44X larger files. CloudFlare is faster for levels 2 to 9. On level 9, the experimental branch outperforms the reference implementation by 2X.
Large corpus
Performance:
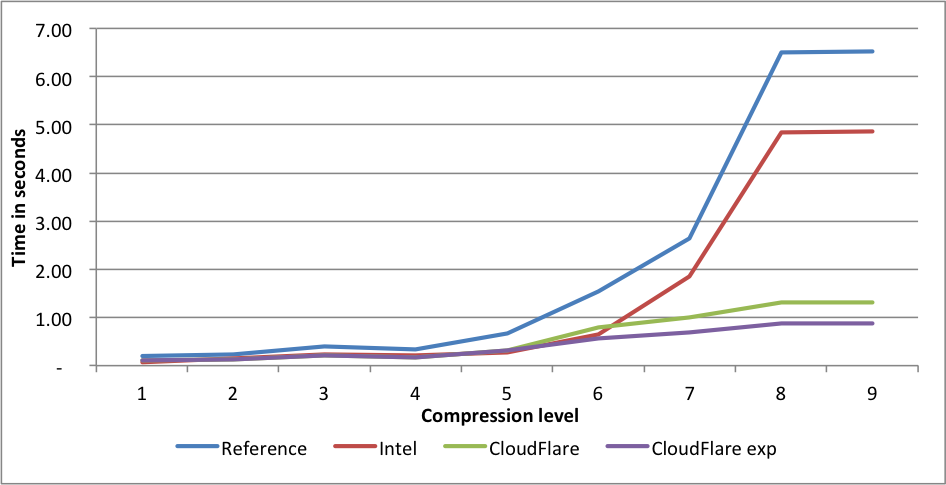 Compression rates:
Compression rates:
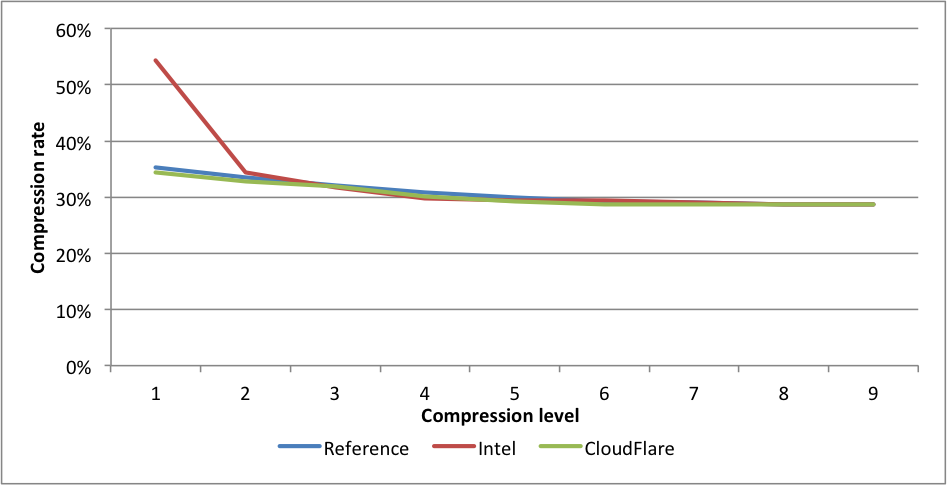 This time Intel is slightly faster for levels 5 and 6 than the CloudFlare implementation. The experimental CloudFlare implementation is faster still on level 6. The compression rate for Intel level 1 is 1.58 lower than CloudFlare. On level 9, the experimental fork is 7.5X(!) faster than reference.
This time Intel is slightly faster for levels 5 and 6 than the CloudFlare implementation. The experimental CloudFlare implementation is faster still on level 6. The compression rate for Intel level 1 is 1.58 lower than CloudFlare. On level 9, the experimental fork is 7.5X(!) faster than reference.
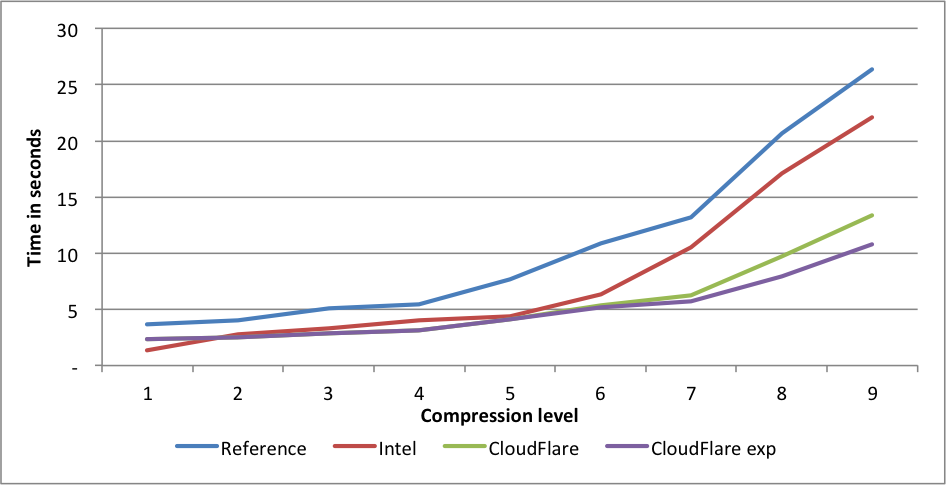
Silesia corpus
Performance:
 Compression rates:
Compression rates:
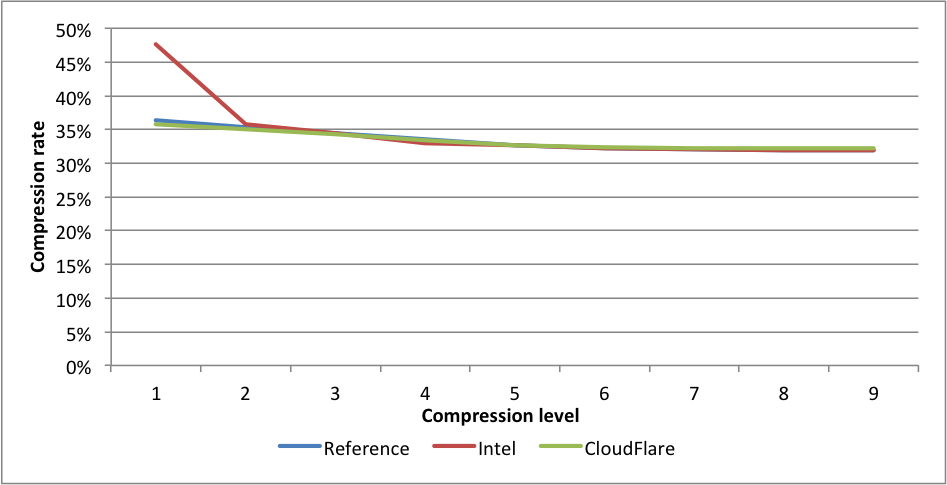 Here again, CloudFlare is the fastest on levels 2 to 9. On level 9 the difference in speed between the experimental fork and the reference fork is 2.44X.
Here again, CloudFlare is the fastest on levels 2 to 9. On level 9 the difference in speed between the experimental fork and the reference fork is 2.44X.
Conclusion
As evident from the benchmarks, the CloudFlare implementation outperforms the competition in the vast majority of settings. We put great effort in making it as fast as possible on our servers.
If you intend to use our library, you should check for yourself if it delivers the best balance of performance and compression for your dataset. As between different file format and sizes performance can vary.
And if you like open source software, don't forget to give back to the community, by contributing your own code!
No really, it's a placebo...
Kaptchuk TJ, Friedlander E, Kelley JM, Sanchez MN, Kokkotou E, Singer JP, Kowalczykowski M, Miller FG, Kirsch I, & Lembo AJ. (2010) Placebos without deception: a randomized controlled trial in irritable bowel syndrome. PloS one, 5(12). PMID: 21203519
Placebos without deception: a randomized controlled trial in irritable bowel syndrome.Cellular Senescence in Regeneration
Eguchi, G., Eguchi, Y., Nakamura, K., Yadav, M., Millán, J., & Tsonis, P. (2011) Regenerative capacity in newts is not altered by repeated regeneration and ageing. Nature Communications, 384. DOI: 10.1038/ncomms1389
Regenerative capacity in newts is not altered by repeated regeneration and ageingYun, M., Davaapil, H., & Brockes, J. (2015) Recurrent turnover of senescent cells during regeneration of a complex structure. eLife. DOI: 10.7554/eLife.05505
Recurrent turnover of senescent cells during regeneration of a complex structurevan Deursen, J. (2014) The role of senescent cells in ageing. Nature, 509(7501), 439-446. DOI: 10.1038/nature13193
The role of senescent cells in ageingSousa-Victor, P., Gutarra, S., García-Prat, L., Rodriguez-Ubreva, J., Ortet, L., Ruiz-Bonilla, V., Jardí, M., Ballestar, E., González, S., Serrano, A.... (2014) Geriatric muscle stem cells switch reversible quiescence into senescence. Nature, 506(7488), 316-321. DOI: 10.1038/nature13013
Geriatric muscle stem cells switch reversible quiescence into senescenceYun, M., Gates, P., & Brockes, J. (2013) Regulation of p53 is critical for vertebrate limb regeneration. Proceedings of the National Academy of Sciences, 110(43), 17392-17397. DOI: 10.1073/pnas.1310519110
Regulation of p53 is critical for vertebrate limb regenerationMuñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M.... (2013) Programmed cell senescence during mammalian embryonic development. Cell, 155(5), 1104-1118. PMID: 24238962
Programmed cell senescence during mammalian embryonic development.Storer, M., Mas, A., Robert-Moreno, A., Pecoraro, M., Ortells, M., Di Giacomo, V., Yosef, R., Pilpel, N., Krizhanovsky, V., Sharpe, J.... (2013) Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell, 155(5), 1119-1130. DOI: 10.1016/j.cell.2013.10.041
Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and PatterningDemaria, M., Ohtani, N., Youssef, S., Rodier, F., Toussaint, W., Mitchell, J., Laberge, R., Vijg, J., Van Steeg, H., Dollé, M.... (2014) An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Developmental Cell, 31(6), 722-733. DOI: 10.1016/j.devcel.2014.11.012
An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AAYou may already be beating cancer
Foxman EF, Storer JA, Fitzgerald ME, Wasik BR, Hou L, Zhao H, Turner PE, Pyle AM, & Iwasaki A. (2015) Temperature-dependent innate defense against the common cold virus limits viral replication at warm temperature in mouse airway cells. Proceedings of the National Academy of Sciences of the United States of America, 112(3), 827-32. PMID: 25561542
Temperature-dependent innate defense against the common cold virus limits viral replication at warm temperature in mouse airway cells.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM.... (2015) Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science (New York, N.Y.), 348(6237), 880-6. PMID: 25999502
Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin.Cherry, J., Liu, B., Frost, J., Lemere, C., Williams, J., Olschowka, J., & O’Banion, M. (2012) Galactic Cosmic Radiation Leads to Cognitive Impairment and Increased Aβ Plaque Accumulation in a Mouse Model of Alzheimer’s Disease. PLoS ONE, 7(12). DOI: 10.1371/journal.pone.0053275
Galactic Cosmic Radiation Leads to Cognitive Impairment and Increased Aβ Plaque Accumulation in a Mouse Model of Alzheimer’s DiseaseInceptionism: Going Deeper into Neural Networks
Update - 13/07/2015
Images in this blog post are licensed by Google Inc. under a Creative Commons Attribution 4.0 International License. However, images based on places by MIT Computer Science and AI Laboratory require additional permissions from MIT for use.
Artificial Neural Networks have spurred remarkable recent progress in image classification and speech recognition. But even though these are very useful tools based on well-known mathematical methods, we actually understand surprisingly little of why certain models work and others don’t. So let’s take a look at some simple techniques for peeking inside these networks.
We train an artificial neural network by showing it millions of training examples and gradually adjusting the network parameters until it gives the classifications we want. The network typically consists of 10-30 stacked layers of artificial neurons. Each image is fed into the input layer, which then talks to the next layer, until eventually the “output” layer is reached. The network’s “answer” comes from this final output layer.
One of the challenges of neural networks is understanding what exactly goes on at each layer. We know that after training, each layer progressively extracts higher and higher-level features of the image, until the final layer essentially makes a decision on what the image shows. For example, the first layer maybe looks for edges or corners. Intermediate layers interpret the basic features to look for overall shapes or components, like a door or a leaf. The final few layers assemble those into complete interpretations—these neurons activate in response to very complex things such as entire buildings or trees.
One way to visualize what goes on is to turn the network upside down and ask it to enhance an input image in such a way as to elicit a particular interpretation. Say you want to know what sort of image would result in “Banana.” Start with an image full of random noise, then gradually tweak the image towards what the neural net considers a banana (see related work in [1], [2], [3], [4]). By itself, that doesn’t work very well, but it does if we impose a prior constraint that the image should have similar statistics to natural images, such as neighboring pixels needing to be correlated.


Indeed, in some cases, this reveals that the neural net isn’t quite looking for the thing we thought it was. For example, here’s what one neural net we designed thought dumbbells looked like:

Instead of exactly prescribing which feature we want the network to amplify, we can also let the network make that decision. In this case we simply feed the network an arbitrary image or photo and let the network analyze the picture. We then pick a layer and ask the network to enhance whatever it detected. Each layer of the network deals with features at a different level of abstraction, so the complexity of features we generate depends on which layer we choose to enhance. For example, lower layers tend to produce strokes or simple ornament-like patterns, because those layers are sensitive to basic features such as edges and their orientations.
 |
| Left: Original photo by Zachi Evenor. Right: processed by Günther Noack, Software Engineer |
 |
| Left: Original painting by Georges Seurat. Right: processed images by Matthew McNaughton, Software Engineer |
{kind=link}


 |
| The original image influences what kind of objects form in the processed image. |
We must go deeper: Iterations
If we apply the algorithm iteratively on its own outputs and apply some zooming after each iteration, we get an endless stream of new impressions, exploring the set of things the network knows about. We can even start this process from a random-noise image, so that the result becomes purely the result of the neural network, as seen in the following images:
 |
| Neural net “dreams”— generated purely from random noise, using a network trained on places by MIT Computer Science and AI Laboratory. See our Inceptionism gallery for hi-res versions of the images above and more (Images marked “Places205-GoogLeNet” were made using this network). |

Topic Pages: a bridge between academia and Wikipedia
Ravenhall M, Škunca N, Lassalle F, & Dessimoz C. (2015) Inferring horizontal gene transfer. PLoS computational biology, 11(5). PMID: 26020646
Inferring horizontal gene transfer.Sunnåker M, Busetto AG, Numminen E, Corander J, Foll M, & Dessimoz C. (2013) Approximate Bayesian computation. PLoS computational biology, 9(1). PMID: 23341757
Approximate Bayesian computation.Liar, Liar: Children with good memories are better liars
Alloway, T., McCallum, F., Alloway, R., & Hoicka, E. (2015) Liar, liar, working memory on fire: Investigating the role of working memory in childhood verbal deception. Journal of Experimental Child Psychology, 30-38. DOI: 10.1016/j.jecp.2015.03.013
Liar, liar, working memory on fire: Investigating the role of working memory in childhood verbal deception