26 Feb 01:57
J. Mater. Chem. A, 2018, 6,4023-4031
DOI: 10.1039/C7TA10976H, Paper
Yueyue Gao, Zhen Wang, Jianqi Zhang, Hong Zhang, Kun Lu, Fengyun Guo, Yulin Yang, Liancheng Zhao, Zhixiang Wei, Yong Zhang
Wide bandgap benzo[1,2-b:4,5-b[prime or minute]]difuran-based polymers for efficient non-fullerene polymer solar cells.
The content of this RSS Feed (c) The Royal Society of Chemistry
26 Feb 01:56
J. Mater. Chem. A, 2018, 6,4572-4589
DOI: 10.1039/C7TA10742K, Review Article
Zhanglin Guo, Liguo Gao, Chu Zhang, Zhenhua Xu, Tingli Ma
We systematically summarized the current progress in low-temperature processed non-TiO2 electron selective layers for perovskite solar cells.
The content of this RSS Feed (c) The Royal Society of Chemistry
26 Feb 01:55
J. Mater. Chem. A, 2018, 6,4179-4188
DOI: 10.1039/C7TA07939G, Paper
Chun-Yu Chang, Chieh-Ping Wang, Rathinam Raja, Leeyih Wang, Cheng-Si Tsao, Wei-Fang Su
Fluorinated PC61BM can be used to fabricate BHJ perovskite films in one step using one solvent in a BHJ precursor solution system.
The content of this RSS Feed (c) The Royal Society of Chemistry
31 Jan 01:01
by Minwoo Nam, Jin Young Huh, Yongkook Park, Yong Cheol Hong, Doo-Hyun Ko
Abstract
Optimizing the interfacial contacts between the photoactive layer and the electrodes is an important factor in determining the performance of organic solar cells (OSCs). A charge-selective layer with tailored electrical properties enhances the charge collection efficiency and interfacial stability. Here, the potential of hydrogenated TiO2 nanoparticles (H-TiO2 NPs) as an efficient electron-selective layer (ESL) material in OSCs is reported for the first time. The H-TiO2 is synthesized by discharge plasma in liquid at atmospheric pressure, which has the benefits of a simple one-pot synthesis process, rapid and mild reaction conditions, and the capacity for mass production. The H-TiO2 exhibits high conductivity and favorable energy level formation for efficient electron extraction, providing a basis for an efficient bilayer ESL system composed of conjugated polyelectrolyte/H-TiO2. Thus, the enhanced charge transport and extraction efficiency with reduced recombination losses at the cathode interfacial contacts is achieved. Moreover, the OSCs composed of H-TiO2 are almost free of light soaking, which has been reported to severely limit the performance and stability of OSCs based on conventional TiO2 ESLs. Therefore, H-TiO2 as a new efficient, stable, and cost-effective ESL material has the potential to open new opportunities for optoelectronic devices.

This study demonstrates the potential of hydrogenated TiO2 (H-TiO2) as an efficient electron-selective layer in optoelectronic devices. The H-TiO2 is simply one-pot mass-produced using a discharge plasma system in liquid at atmospheric pressure. The H-TiO2 exhibits high conductivity and favorable energy level formation, resulting in the high-efficiency and light-soaking-free organic solar cells.
31 Jan 01:00
by Wenzhan Xu, Luyao Zheng, Xiaotao Zhang, Yu Cao, Tianyu Meng, Dezhen Wu, Lei Liu, Wenping Hu, Xiong Gong
Abstract
In the past years, hybrid perovskite materials have attracted great attention due to their superior optoelectronic properties. In this study, the authors report the utilization of cobalt (Co2+) to partially substitute lead (Pb2+) for developing novel hybrid perovskite materials, CH3NH3Pb1-xCoxI3 (where x is nominal ratio, x = 0, 0.1, 0.2 and 0.4). It is found that the novel perovskite thin films possess a cubic crystal structure with superior thin film morphology and larger grain size, which is significantly different from pristine thin film, which possesses the tetragonal crystal structure, with smaller grain size. Moreover, it is found that the 3d orbital of Co2+ ensures higher electron mobilities and electrical conductivities of the CH3NH3Pb1-xCoxI3 thin films than those of pristine CH3NH3Pb4 thin film. As a result, a power conversion efficiency of 21.43% is observed from perovskite solar cells fabricated by the CH3NH3Pb0.9Co0.1I3 thin film. Thus, the utilization of Co, partially substituting for Pb to tune physical properties of hybrid perovskite materials provides a facile way to boost device performance of perovskite solar cells.

The utilization of cobalt (Co2+) to partially substitute lead (Pb2+) for developing novel hybrid perovskite materials and perovskite solar cells is reported. A power conversion efficiency of 21.43% is observed from perovskite solar cells fabricated by the CH3NH3Pb0.9Co0.1I3 thin film, which is due to it possessing a cubic crystal structure with superior thin film morphology and larger grain size.
31 Jan 00:56
by Xinqian Zhang, Gang Wu, Weifei Fu, Minchao Qin, Weitao Yang, Jielin Yan, Zhongqiang Zhang, Xinhui Lu, Hongzheng Chen
Abstract
Increasing the power conversion efficiency (PCE) of the two-dimensional (2D) perovskite-based solar cells (PVSCs) is really a challenge. Vertical orientation of the 2D perovskite film is an efficient strategy to elevate the PCE. In this work, vertically orientated highly crystalline 2D (PEA)2(MA)n–1PbnI3n+1 (PEA= phenylethylammonium, MA = methylammonium, n = 3, 4, 5) films are fabricated with the assistance of an ammonium thiocyanate (NH4SCN) additive by a one-step spin-coating method. Planar-structured PVSCs with the device structure of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate)/(PEA)2(MA)n–1PbnI3n+1/[6,6]-phenyl-C61-butyric acid methyl ester/bahocuproine/Ag are fabricated. The PCE of the PVSCs is boosted from the original 0.56% (without NH4SCN) to 11.01% with the optimized NH4SCN addition at n = 5, which is among the highest PCE values for the low-n (n < 10) 2D perovskite-based PVSCs. The improved performance is attributed to the vertically orientated highly crystalline 2D perovskite thin films as well as the balanced electron/hole transportation. The humidity stability of this oriented 2D perovskite thin film is also confirmed by the almost unchanged X-ray diffraction patterns after 28 d exposed to the moisture in a humidity-controlled cabinet (Hr = 55 ± 5%). The unsealed device retains 78.5% of its original PCE after 160 h storage in air atmosphere with humidity of 55 ± 5%. The results provide an effective approach toward a highly efficient and stable PVSC for future commercialization.

The advantage of phenylethylammonium (PEA+) in forming pinhole-free 2D (PEA)2(methylammonium (MA))n−1PbnI3n+1 n = 3, 4, 5) perovskite film with vertical orientation and high crystallinity under assistance of an ammonium thiocyanate additive by one-step spin-coating method is demonstrated. The optimized planar-structured perovskite solar cell based on vertically oriented (PEA)2(MA)4Pb5I16 (n = 5) film presents the best power conversion efficiency of 11.01% with excellent stability.
31 Jan 00:56
by Yanbo Wang, Yamin Zhang, Nailiang Qiu, Huanran Feng, Huanhuan Gao, Bin Kan, Yanfeng Ma, Chenxi Li, Xiangjian Wan, Yongsheng Chen
Abstract
Three acceptor–donor–acceptor type nonfullerene acceptors (NFAs), namely, F–F, F–Cl, and F–Br, are designed and synthesized through a halogenation strategy on one successful nonfullerene acceptor FDICTF (F–H). The three molecules show red-shifted absorptions, increased crystallinities, and higher charge mobilities compared with the F–H. After blending with donor polymer PBDB-T, the F–F-, F–Cl-, and F–Br-based devices exhibit power conversion efficiencies (PCEs) of 10.85%, 11.47%, and 12.05%, respectively, which are higher than that of F–H with PCE of 9.59%. These results indicate that manipulating the absorption range, crystallinity and mobilities of NFAs by introducing different halogen atoms is an effective way to achieve high photovoltaic performance, which will offer valuable insight for the designing of high-efficiency organic solar cells.

Through a halogenation strategy onto the end-capping group in the FDICTF-based small-molecule acceptor, red-shifted absorptions, increased crystallinities, and higher charge mobilities are achieved. The device based on F–Br with power conversion efficiency of 12.05% and remarkable FF of 76% is one of only a few organic solar cells with efficiencies over 12% reported to date.
30 Jan 06:37
by Amaia Diaz Zerio, Christian Müller
Abstract
The stability of donor:acceptor (D:A) semiconductor blends plays a key role in the development of solution-processed organic solar cells. One essential condition for both high-yield production and a long lifetime is excellent thermal stability. Recently, A1:A2 acceptor mixtures have received considerable attention and alloys of two miscible acceptors are singled out as a powerful tool for the design of efficient and durable organic solar cells. This progress report introduces a thermodynamic rationale for the superior thermal stability and reproducibility that is observed for some ternary blends. The increase in entropy upon mixing of several acceptors reduces the tendency for phase separation as well as crystallization, which facilitates the controlled formation of a fine blend nanostructure. Further, when combined with a high glass transition temperature many ternary blends can be readily quenched into a glassy state. Recent progress with regard to the thermal stability and efficiency of D:A1:A2 ternary blends is summarized in the light of the thermodynamic and kinetic arguments discussed in this article. Both, fullerene and fullerene-free acceptor alloys now yield solar cell efficiencies in excess of 10%, which indicates that ternary blends are a promising avenue that is poised to considerably enhance the prospect of organic photovoltaics.

Ternary organic solar cells display superior thermal stability and reproducibility. The increase in entropy upon mixing of several acceptors reduces the tendency for phase separation and crystallization. When combined with a high glass transition temperature many ternary blends can be quenched into a glassy state. Both, fullerene and fullerene-free acceptor alloys now yield solar cell efficiencies in excess of 10%.
30 Jan 06:35
by Nicola Gasparini, Andrew Wadsworth, Maximilian Moser, Derya Baran, Iain McCulloch, Christoph J. Brabec
Abstract
Organic bulk heterojunction solar cells based on small molecule acceptors have recently seen a rapid rise in the power conversion efficiency with values exceeding 13%. This impressive achievement has been obtained by simultaneous reduction of voltage and charge recombination losses within this class of materials as compared to fullerene-based solar cells. In this contribution, the authors review the current understanding of the relevant photophysical processes in highly efficient nonfullerene acceptor (NFA) small molecules. Charge generation, recombination, and charge transport is discussed in comparison to fullerene-based composites. Finally, the authors review the superior light and thermal stability of nonfullerene small molecule acceptor based solar cells, and highlight the importance of NFA-based composites that enable devices without early performance loss, thus resembling so-called burn-in free devices.

In this contribution, the authors review the current understanding of the relevant photophysical processes, as well as the superior light and thermal stability of efficient nonfullerene acceptor small molecules in comparison to fullerenes-based composites.
30 Jan 01:34
by Yue Hu, Zhihui Zhang, Anyi Mei, Youyu Jiang, Xiaomeng Hou, Qifei Wang, Kai Du, Yaoguang Rong, Yinhua Zhou, Gengzhao Xu, Hongwei Han
Abstract
A bifunctional conjugated organic molecule 4-(aminomethyl) benzoic acid hydroiodide (AB) is designed and employed as an organic cation in organic–inorganic halide perovskite materials. Compared with the monofunctional cation benzylamine hydroiodide (BA) and the nonconjugated bifunctional organic molecule 5-ammonium valeric acid, devices based on AB-MAPbI3 show a good stability and a superior power conversion efficiency of 15.6% with a short-circuit current of 23.4 mA cm−2, an open-circuit voltage of 0.94 V, and a fill factor of 0.71. The bifunctional conjugated cation not only benefits the growth of perovskite crystals in the mesoporous network, but also facilitates the charge transport. This investigation helps explore new approaches to rational design of novel organic cations for perovskite materials.

A bifunctional conjugated organic molecule AB is designed and employed as an organic cation in perovskite materials. Compared with the monofunctional cation BA and the nonconjugated bifunctional cation AVA, the devices based on AB-MAPbI3 show superior efficiency with good stability. The bifunctional-conjugated cation not only benefits the growth of perovskite crystals in the mesoporous network, but also facilitates the charge transport.
27 Jan 02:34
by Guangchao Han, Yuanping Yi, Zhigang Shuai
Abstract
Molecular packing structures in the active layers have a crucial impact on the electronic processes for organic solar cells. To date, however, it is still difficult to probe molecular self-assembling and packing structures at the atomic level by experimental techniques, which is hindering reliable understanding of the structure–property relationship. Accordingly, theoretical simulations provide a useful tool and are becoming more and more important. Here, recent advances in theoretical simulations for organic solar cells are reviewed. First, a brief introduction of theoretical methodologies, including the strategies of molecular dynamics simulations of active-layer processing procedures and quantum-chemical methods for calculating electron transfer processes, is given. Then, the influences of molecular packing structures on charge generation, charge recombination, and charge transport are analyzed and discussed from a theoretical perspective. Finally, prospects and challenges are pointed out for theoretical prediction of the electrical characteristics and photoelectric conversion efficiencies of organic solar cells from molecular structures.

Recent advances in theoretical simulations for organic solar cells are summarized, ranging from molecular packing structures to electronic processes. Insights into the correlation between molecular structures, molecular packing morphologies, and electronic processes are provided, which would be helpful to molecular design toward improving photovoltaic performance.
27 Jan 02:34
by Arman Mahboubi Soufiani, Jincheol Kim, Anita Ho-Baillie, Martin Green, Ziv Hameiri
Abstract
In this essay, the authors use two properly encapsulated high-efficiency mesoscopic perovskite solar cells (PSCs), which use a state-of-the-art perovskite composition (HC(NH2)2PbI3)0.85(CH3NH3PbBr3)0.15 with excess PbI2 as the active layer, to demonstrate the potential effect of dynamical electroluminescence responses on the analysis and interpretation of PSCs electrical characteristic. The essay does not aim to determine how to overcome this issue, nor to investigate its physical/chemical origin, although tentative propositions are made; but rather, to warn researchers in the field about the interpretation and reporting the results obtained from luminescence imaging measurements and the effect of image collection timing on the results. This is a critical message since the authors predict that luminescence imaging techniques will soon become one of the key tools for PSCs characterization, both for long-term stability assessment and fabrication process optimization.

It is expected that luminescence imaging will become a key tool for perovskite solar cells (PSCs) stability assessment and fabrication process optimization, especially for large-area devices. Even state-of-the-art mesoscopic PSCs, with a small photocurrent hysteresis, show dynamic electroluminescence signal, which complicates immediate and accurate analysis of luminescence imaging measurements results. This article demonstrates this while focusing on the effect of image collection timing.
27 Jan 02:34
by Han-Hee Cho, Seonha Kim, Taesu Kim, Vijaya Gopalan Sree, Sung-Ho Jin, Felix Sunjoo Kim, Bumjoon J. Kim

In article number 1701436 by Bumjoon J. Kim and co-workers, a series of naphthalenediimide-based polymer acceptors with superior electron mobility and large dipole moment difference is developed by incorporating electron-withdrawing cyanovinylene groups into a polymer backbone. All-polymer solar cells based on these polymers generate outstanding power conversion efficiency of 7.4% with high fill factor (65%), by virtue of the high electron transport and efficient exciton dissociation with greatly suppressed charge recombination.
27 Jan 02:34
by Yinghong Hu, Eline M. Hutter, Philipp Rieder, Irene Grill, Jonas Hanisch, Meltem F. Aygüler, Alexander G. Hufnagel, Matthias Handloser, Thomas Bein, Achim Hartschuh, Kristofer Tvingstedt, Vladimir Dyakonov, Andreas Baumann, Tom J. Savenije, Michiel L. Petrus, Pablo Docampo
Abstract
Adding cesium (Cs) and rubidium (Rb) cations to FA0.83MA0.17Pb(I0.83Br0.17)3 hybrid lead halide perovskites results in a remarkable improvement in solar cell performance, but the origin of the enhancement has not been fully understood yet. In this work, time-of-flight, time-resolved microwave conductivity, and thermally stimulated current measurements are performed to elucidate the impact of the inorganic cation additives on the trap landscape and charge transport properties within perovskite solar cells. These complementary techniques allow for the assessment of both local features within the perovskite crystals and macroscopic properties of films and full devices. Strikingly, Cs-incorporation is shown to reduce the trap density and charge recombination rates in the perovskite layer. This is consistent with the significant improvements in the open-circuit voltage and fill factor of Cs-containing devices. By comparison, Rb-addition results in an increased charge carrier mobility, which is accompanied by a minor increase in device efficiency and reduced current–voltage hysteresis. By mixing Cs and Rb in quadruple cation (Cs-Rb-FA-MA) perovskites, the advantages of both inorganic cations can be combined. This study provides valuable insights into the role of these additives in multiple-cation perovskite solar cells, which are essential for the design of high-performance devices.

Time-resolved microwave conductivity, time-of-flight, and thermally stimulated current measurements reveal that Cs reduces the trap density in hybrid lead halide perovskites. Rb additives enhance the charge carrier mobility, but show minor effects on the trap landscape. The increase in open-circuit voltage in multiple-cation perovskite solar cells can be related to a reduced trap density through Cs-incorporation.
27 Jan 02:31
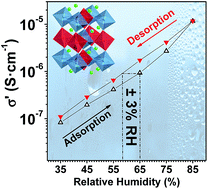
J. Mater. Chem. A, 2018, 6,5430-5442
DOI: 10.1039/C7TA09496E, Paper
C. de la Torre-Gamarra, M. Woszczak, B. Levenfeld, A. Varez, E. Garcia-Gonzalez, E. Urones-Garrote, V. Di Noto
The electrical response to RH% of a new La2LiNbO6 double perovskite as a sensing material in a humidity sensor.
The content of this RSS Feed (c) The Royal Society of Chemistry
26 Jan 01:18
J. Mater. Chem. A, 2018, 6,3649-3658
DOI: 10.1039/C7TA10017E, Paper
Zhiqiang Liang, Wenkai Liang, Weijing Shao, Jing Huang, Tianfu Guan, Peng Wen, Guozhong Cao, Lin Jiang
Aluminum nanodisk arrays on TiO2 thin films were fabricated via a new self-assembly nanoparticle template method, and successfully employed to enhance the photovoltaic performance of organic photovoltaics for the first time.
The content of this RSS Feed (c) The Royal Society of Chemistry
26 Jan 01:16
by Jin Soo Kang, Jae-Yup Kim, Jungjin Yoon, Jin Kim, Jiwoong Yang, Dong Young Chung, Min-cheol Kim, Hansol Jeong, Yoon Jun Son, Bong Gyu Kim, Juwon Jeong, Taeghwan Hyeon, Mansoo Choi, Min Jae Ko, Yung-Eun Sung
Abstract
Organic/inorganic hybrid solar cells, typically mesoscopic and perovskite solar cells, are regarded as promising candidates to replace conventional silicon or thin film photovoltaics. There have been intensive investigations on the development of advanced materials for improved power conversion efficiencies, however, economical feasibilities and reliabilities of the organic/inorganic photovoltaics are yet to reach at a sufficient level for practical utilizations. In this study, cobalt nitride (CoN) nanofilms prepared by room-temperature vapor deposition in an inert N2 atmosphere, which is a facile and highly reproducible procedure, are proposed as a low-cost counter electrode in mesoscopic dye-sensitized solar cells (DSCs) and a hole transport material in inverted planar perovskite solar cells (PSCs) for the first time. The CoN film successfully replaces conventional Pt in DSCs, resulting in a power conversion efficiency comparable to the ones based on Pt. In addition, PSCs employing the CoN manifest high efficiency even up to 15.0%, which is comparable to state-of-the-art performance in the cases of PSCs employing inorganic hole transporters. Furthermore, flexible solar cell applications of the CoN are performed in both mesoscopic and perovskite solar cells, verifying the advantages of the room-temperature deposition process and feasibilities of the CoN nanofilms in various fields.

CoN nanofilms prepared by room-temperature vapor deposition are applied as electrocatalysts and hole transport materials in organic/inorganic hybrid solar cells. The CoN counter electrode in place of Pt manifests a comparably high performance, and power conversion efficiency achieved in perovskite solar cells employing the CoN hole transporter is among the state-of-the-art results from inorganic hole transport materials.
25 Jan 01:03
by Randi Azmi, Chang-Lyoul Lee, In Hwan Jung, Sung-Yeon Jang
Abstract
In most current state-of-the-art perovskite solar cells (PSCs), high-temperature (≈500 °C)-sintered metal oxides are employed as electron-transporting layers (ETLs). To lower the device processing temperature, the development of low-temperature-processable ETL materials (such as solution-processed ZnO) has received growing attention. However, thus far, the use of solution-processed ZnO is limited because the reverse decomposition reaction that occurs at ZnO/perovskite interfaces significantly degrades the charge collection and stability of PSCs. In this work, the reverse decomposition reaction is successfully retarded by sulfur passivation of solution-processed ZnO. The sulfur passivation of ZnO by a simple chemical means, efficiently reduces the oxygen-deficient defects and surface oxygen-containing groups, thus effectively preventing reverse decomposition reactions during and after formation of the perovskite active layers. Using the low-temperature-processed sulfur-passivated ZnO (ZnO–S), perovskite layers with higher crystallinity and larger grain size are obtained, while the charge extraction at the ZnO/perovskite interface is significantly improved. As a result, the ZnO–S-based PSCs achieve substantially improved power-conversion-efficiency (PCE) (19.65%) and long-term air-storage stability (90% retention after 40 d) compared with pristine ZnO-based PSCs (16.51% and 1% retention after 40 d). Notably, the PCE achieved is the highest recorded (19.65%) for low-temperature ZnO-based PSCs.

Air-stable high efficiency perovskite solar cells are developed using sulfur-passivated ZnO electron transport layers. Sulfur passivation of ZnO effectively prevents the interfacial reverse reaction from perovskite to PbI2, while the surface hydrophobicity of ZnO is increased. The results show that the quality of perovskite layers is improved and the interfacial charge recombination is reduced.
25 Jan 01:03
by Jianyu Yuan, Michael J. Ford, Yalong Xu, Yannan Zhang, Guillermo C. Bazan, Wanli Ma
Abstract
All-polymer solar cells (all-PSCs) are attractive as alternatives to fabricate thermally and mechanically stable solar cells, especially with recent improvements in their power conversion efficiency (PCE). In this work, efficient all-PSCs with near-infrared response (up to 850 nm) are developed using newly designed regioregular polymer donors with relatively narrow optical gap. These all-PSCs systems achieve PCEs up to 6.0% after incorporating fluorine into the polymer backbone. More importantly, these polymers exhibit absorbance that is complementary to previously reported wide bandgap polymer donors. Thus, the superior properties of the newly designed polymers afford opportunities to fabricate the first spectrally matched all-polymer tandem solar cells with high performance. A PCE of 8.3% is then demonstrated which is the highest efficiency so far for all-polymer tandem solar cells. The design of narrow bandgap polymers provides new directions to enhance the PCE of emerging single-junction and tandem all polymer solar cells.

By adopting D1-A-D2-A ternary structure, a pair of novel regioregular polymers, namely PBBSB and PBFSF, are synthesized. Benefiting from the new arrangement and molecular fluorination, the polymer exhibits relatively narrow optical gap, good intermolecular packing, and excellent charge transport. More importantly, it is shown that these functional donor polymers can achieve high efficiency in either single-junction or tandem all-polymer solar cells.
25 Jan 01:01
by Jingru Zhang, Dongliang Bai, Zhiwen Jin, Hui Bian, Kang Wang, Jie Sun, Qian Wang, Shengzhong (Frank) Liu
Abstract
All-inorganic CsPbBrI2 perovskite has great advantages in terms of ambient phase stability and suitable band gap (1.91 eV) for photovoltaic applications. However, the typically used structure causes reduced device performance, primarily due to the large recombination at the interface between the perovskite, and the hole-extraction layer (HEL). In this paper, an efficient CsPbBrI2 perovskite solar cell (PSC) with a dimensionally graded heterojunction is reported, in which the CsPbBrI2 material is distributed within bulk–nanosheet–quantum dots or 3D–2D–0D dimension-profiled interface structure so that the energy alignment is optimized in between the valence and conduction bands of both CsPbBrI2 and the HEL layers. Specifically, the valence-/conduction-band edge is leveraged to bend with synergistic advantages: the graded combination enhances the hole extraction and conduction efficiency with effectively decreased recombination loss during the hole-transfer process, leading to an enhanced built-in electric field, hence a high VOC of as much as 1.19 V. The profiled structure induces continuously upshifted energy levels, resulting in a higher JSC of as much as 12.93 mA cm−2 and fill factor as high as 80.5%, and therefore record power conversion efficiency (PCE) of 12.39%. As far as it is known, this is the highest PCE for CsPbBrI2 perovskite-based PSC.

Here, a 3D–2D–0D multi-graded interface based on CsPbBrI2 bulk, nanosheets, and quantum dots is first designed for CsPbBrI2 perovskite solar cells. Such a multigraded surface favorably reduces the recombination at the CsPbBrI2/poly[bis(4-phenyl) (2,4,6-trimethylphenyl)amine] interface, resulting in a record stabilized power conversion efficiency of 12.39%, a nearly 20% increase compared with 10.38% for ungraded devices.
25 Jan 01:00
by Jiangzhao Chen, Ja-Young Seo, Nam-Gyu Park
Abstract
To solve the stability issues of perovskite solar cells (PSC), here a novel interface engineering strategy that a versatile ultrathin 2D perovskite (5-AVA)2PbI4 (5-AVA = 5-ammoniumvaleric acid) passivation layer that is in situ incorporated at the interface between (FAPbI3)0.88(CsPbBr3)0.12 and the hole transporting CuSCN is reported. Surface analysis using X-ray photoelectron spectroscopy confirms the formation of 2D perovskite. Hysteresis is reduced by the interfacial 2D layer, which could be ascribed to improvement of interfacial charge extraction efficiency, associated with suppression of recombination. Moreover, introduction of the interface passivating layer enhances the moisture stability and photostability as compared to the control perovskite film due to hydrophobic nature of 2D perovskite. The unencapsulated device retains 98% of the initial power conversion efficiency (PCE) after 63 d under moisture exposure of about 10% in the dark. A PCE of the control device is boosted from 13.72 to 16.75% as a consequence of enhanced open-circuit voltage (Voc) and fill factor along with slightly increased short-circuit current density (Jsc), which results from reduced trap states of (FAPbI3)0.88(CsPbBr3)0.12 as evidenced by enhanced carrier lifetimes and charge extraction. The perovskite/hole transport material interface engineering gives insight into simultaneous improvements of PCE and device stability.

A versatile ultrathin 2D perovskite (5-AVA)2PbI4 (5-AVA = 5-ammoniumvaleric acid) interlayer is in situ incorporated at the back contact interface between perovskite and CuSCN, which reduces current–voltage hysteresis and improves simultaneously power conversion efficiency (PCE) and stability. An unencapsulated device retains 98% of the initial PCE after 63 days under 10% relative humidity in the dark.
25 Jan 00:59
by Mohammad Mahdi Tavakoli, Shaik Mohammed Zakeeruddin, Michael Grätzel, Zhiyong Fan
Abstract
Fast research progress on lead halide perovskite solar cells has been achieved in the past a few years. However, the presence of lead (Pb) in perovskite composition as a toxic element still remains a major issue for large-scale deployment. In this work, a novel and facile technique is presented to fabricate tin (Sn)-rich perovskite film using metal precursors and an alloying technique. Herein, the perovskite films are formed as a result of the reaction between Sn/Pb binary alloy metal precursors and methylammonium iodide (MAI) vapor in a chemical vapor deposition process carried out at 185 °C. It is found that in this approach the Pb/Sn precursors are first converted to (Pb/Sn)I2 and further reaction with MAI vapor leads to the formation of perovskite films. By using Pb–Sn eutectic alloy, perovskite films with large grain sizes up to 5 µm can be grown directly from liquid phase metal. Consequently, using an alloying technique and this unique growth mechanism, a less-toxic and efficient perovskite solar cell with a power conversion efficiency (PCE) of 14.04% is demonstrated, while pure Sn and Pb perovskite solar cells prepared in this manner yield PCEs of 4.62% and 14.21%, respectively. It is found that this alloying technique can open up a new direction to further explore different alloy systems (binary or ternary alloys) with even lower melting point.

Sn-rich perovskite solar cells with large grains are fabricated from a Pb–Sn eutectic alloy in the liquid phase by using a chemical vapor deposition technique, resulting in a device power conversion efficiency of 14.04%, which is comparable with that of pure Pb devices and among the highest for Sn-rich binary Sn/Pb metal perovskite solar cells.
24 Jan 06:31
J. Mater. Chem. A, 2018, 6,3724-3729
DOI: 10.1039/C7TA10026D, Paper
Ran Hou, Miao Li, Shiyu Feng, Yahui Liu, Liangliang Wu, Zhaozhao Bi, Xinjun Xu, Wei Ma, Zhishan Bo
A fused pentacyclic small molecule acceptor with cis-arranged alkyl side chains was firstly synthesized and applied in polymer solar cells.
The content of this RSS Feed (c) The Royal Society of Chemistry
24 Jan 06:20
by Chang Woo Myung, Jeonghun Yun, Geunsik Lee, Kwang S. Kim
Abstract
As the race toward higher efficiency for inorganic/organic hybrid perovskite solar cells (PSCs) is becoming highly competitive, a design scheme to maximize carrier transport toward higher power efficiency has been urgently demanded. In this study, a hidden role of A-site cations of PSCs in carrier transport, which has been largely neglected is unraveled, i.e., tuning the Fröhlich electron–phonon (e–ph) coupling of longitudinal optical (LO) phonon by A-site cations. The key for steering Fröhlich polaron is to control the interaction strength and the number of proton (or lithium) coordination to halide ions. The coordination to I− alleviates electron–phonon scattering by either decreasing the Born effective charge or absorbing the LO motion of I. This novel principle discloses low electron–phonon coupling in several promising organic cations including hydroxyl–ammonium cation (NH3OH+), hydrazinium cation (NH3NH2+) and possibly Li+ solvating methylamine (Li+∙∙∙NH2CH3), on a par with methyl–ammonium cations. A new perspective on the role of A-site cations could help in improving power efficiency and accelerating the application of PSCs.

A hidden role of A-site cations in perovskite solar cells in steering Fröhlich polaron coupling is disclosed. Design principles suggest that A-site cations need to be close to halides and to maximize the coordination to halides. Based on first principles and many-body theory, organic cations such as NH3OH+, LiNH2CH3+, and NH3F+ are predicted to be promising.
24 Jan 06:20
by Andreas Kunzmann, Silvia Valero, Ángel E. Sepúlveda, Marisa Rico-Santacruz, Elena Lalinde, Jesús R. Berenguer, Javier García-Martínez, Dirk M. Guldi, Elena Serrano, Rubén D. Costa
Abstract
In this work, a new strategy to design low-temperature (≤200 °C) sintered dye-sensitized solar cells (lt-DSSC) is reported to enhance charge collection efficiencies (ηcoll), photoconversion efficiencies (η), and stabilities under continuous operation conditions. Realization of lt-DSSC is enabled by the integration of hybrid nanoparticles based on TiO2-Ru(II) complex (TiO2_Ru_IS)—obtained by in situ bottom-up construction of Ru(II) N3 dye-sensitized titania—into the photoelectrode. Incentives for the use of TiO2_Ru_IS are i) dye stability due to its integration into the TiO2 anatase network and ii) enhanced charge collection yield due to its significant resistance toward electron recombination with electrolytes. It is demonstrated that devices with single-layer photoelectrodes featuring blends of P25 and TiO2_Ru_IS give rise to a 60% ηcoll relative to a 46% ηcoll for devices with P25-based photoelectrodes. Responsible for this trend is a better charge transport and a reduced electron recombination. When using a multilayered photoelectrode architecture with a top layer based only on TiO2_Ru_IS, devices with an even higher ηcoll (74%) featuring a η of around 8.75% and stabilities of 600 h are achieved. This represents the highest values reported for lt-DSSC to date.

New dye-titania hybrid nanoparticles, namely TiO2_IS_Ru, consisting of the in situ incorporation of a Ru(II) dye inside the structure of the anatase nanoparticles during their synthesis are utilized for designing low temperature sintered dye-sensitized solar cells with high stability and unprecedented efficiencies.
24 Jan 06:19
by Hua Zhang, Huan Wang, Hongmei Zhu, Chu-Chen Chueh, Wei Chen, Shihe Yang, Alex K.-Y. Jen
Abstract
Organic–inorganic hybrid perovskite solar cells (PVSCs) have become the front-running photovoltaic technology nowadays and are expected to profoundly impact society in the near future. However, their practical applications are currently hampered by the challenges of realizing high performance and long-term stability simultaneously. Herein, the development of inverted PVSCs is reported based on low temperature solution-processed CuCrO2 nanocrystals as a hole-transporting layer (HTL), to replace the extensively studied NiOx counterpart due to its suitable electronic structure and charge carrier transporting properties. A ≈45 nm thick compact CuCrO2 layer is incorporated into an inverted planar configuration of indium tin oxides (ITO)/c-CuCrO2/perovskite/[6,6]-phenyl-C61-butyric acid methyl ester (PCBM)/bathocuproine (BCP)/Ag, to result in the high steady-state power conversion efficiency of 19.0% versus 17.1% for the typical low temperature solution-processed NiOx-based devices. More importantly, the optimized CuCrO2-based device exhibits a much enhanced photostability than the reference device due to the greater UV light-harvesting of the CuCrO2 layer, which can efficiently prevent the perovskite film from intense UV light exposure to avoid associated degradation. The results demonstrate the promising potential of CuCrO2 nanocrystals as an efficient HTL for realizing high-performance and photostable inverted PVSCs.

A new, multifunctional CuCrO2 hole-transporting layer is developed and incorporated into the inverted perovskite solar cells. The CuCrO2 layer with superior electronic and optical properties is proved to not only enhance the photovoltaic performance but also improve the device photostability via the UV-blocking effect. Consequently, a high power conversion efficiency of 19.0% with enhanced device photostability is successfully demonstrated.
24 Jan 06:19
by Guangjun Nan, Xu Zhang, Mojtaba Abdi-Jalebi, Zahra Andaji-Garmaroudi, Samuel D. Stranks, Gang Lu, David Beljonne
Abstract
Lead tri-iodide methylammonium (MAPbI3) perovskite polycrystalline materials show complex optoelectronic behavior, largely because their 3D semiconducting inorganic framework is strongly perturbed by the organic cations and ubiquitous structural or chemical inhomogeneities. Here, a newly developed time-dependent density functional theory-based theoretical formalism is taken advantage of. It treats electron–hole and electron–nuclei interactions on the same footing to assess the many-body excited states of MAPbI3 perovskites in their pristine state and in the presence of point chemical defects. It is shown that lead and iodine vacancies yield deep trap states that can be healed by dynamic effects, namely rotation of the methylammonium cations in response to point charges, or through slight changes in chemical composition, namely by introducing a tiny amount of chlorine dopants in the defective MAPbI3. The theoretical results are supported by photoluminescence experiments on MAPbI3−mClm and pave the way toward the design of defect-free perovskite materials with optoelectronic performance approaching the theoretical limits.

How methylammonium cations and chlorine dopants heal defects in lead tri-iodide methylammonium (MAPbI3) perovskites is proposed. Time-dependent density functional theory excited-state calculations in defective MAPbI3 show crossovers from confined to extended states when varying the electrostatic environment around vacancies. Deep trap states are dynamically healed through collective rotation of the methylammonium cations and by introducing tiny amounts of chlorine dopants.
24 Jan 06:18
by Yuanbao Lin, Yingzhi Jin, Sheng Dong, Wenhao Zheng, Junyu Yang, Alei Liu, Feng Liu, Yufeng Jiang, Thomas P. Russell, Fengling Zhang, Fei Huang, Lintao Hou
Abstract
The current work reports a high power conversion efficiency (PCE) of 9.54% achieved with nonfullerene organic solar cells (OSCs) based on PTB7-Th donor and 3,9-bis(2-methylene-(3-(1,1-dicyanomethylene)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene) (ITIC) acceptor fabricated by doctor-blade printing, which has the highest efficiency ever reported in printed nonfullerene OSCs. Furthermore, a high PCE of 7.6% is realized in flexible large-area (2.03 cm2) indium tin oxide (ITO)-free doctor-bladed nonfullerene OSCs, which is higher than that (5.86%) of the spin-coated counterpart. To understand the mechanism of the performance enhancement with doctor-blade printing, the morphology, crystallinity, charge recombination, and transport of the active layers are investigated. These results suggest that the good performance of the doctor-blade OSCs is attributed to a favorable nanoscale phase separation by incorporating 0.6 vol% of 1,8-diiodooctane that prolongs the dynamic drying time of the doctor-bladed active layer and contributes to the migration of ITIC molecules in the drying process. High PCE obtained in the flexible large-area ITO-free doctor-bladed nonfullerene OSCs indicates the feasibility of doctor-blade printing in large-scale fullerene-free OSC manufacturing. For the first time, the open-circuit voltage is increased by 0.1 V when 1 vol% solvent additive is added, due to the vertical segregation of ITIC molecules during solvent evaporation.

Printed nonfullerene organic solar cells are investigated with a power conversion efficiency of 9.54% via incorporating a 1,8-diiodooctane additive for achieving a favorable nanoscale phase separation. The migration of nonfullerene acceptor molecules from bottom to top helps form the optimal donor/acceptor interface distribution, leading to the reduced exciton recombination and optimized electrical parameters.
24 Jan 05:40
by Mahshid Ahmadi, Liam Collins, Alexander Puretzky, Jia Zhang, Jong Kahk Keum, Wei Lu, Ilia Ivanov, Sergei V. Kalinin, Bin Hu
Abstract
Organometallic halide perovskites (OMHPs) have attracted broad attention as prospective materials for optoelectronic applications. Among the many anomalous properties of these materials, of special interest are the ferroelectric properties including both classical and relaxor-like components, as a potential origin of slow dynamics, field enhancement, and anomalous mobilities. Here, ferroelectric properties of the three representative OMHPs are explored, including FAPbxSn1–xI3 (x = 0, x = 0.85) and FA0.85MA0.15PbI3 using band excitation piezoresponse force microscopy and contact mode Kelvin probe force microscopy, providing insight into long- and short-range dipole and charge dynamics in these materials and probing ferroelectric density of states. Furthermore, second-harmonic generation in thin films of OMHPs is observed, providing a direct information on the noncentrosymmetric polarization in such materials. Overall, the data provide strong evidence for the presence of ferroelectric domains in these systems; however, the domain dynamics is suppressed by fast ion dynamics. These materials hence present the limit of ferroelectric materials with spontaneous polarization dynamically screened by ionic and electronic carriers.

Anomalous ferroelectric properties are reported in a series of organometallic halide perovskite compounds. Noncentrosymmetric polarization detected from second-harmonic generation provides a precondition for intrinsic polarization. A classical ferroelectric domain with difference in amplitude and phase is observed, and there are no frequency changes in band excitation piezoresponse force microscopy, ruling out topographic and microstructural artefacts. Finally, Kelvin probe force microscopy reveals that the ferroelectricity is hidden by ion migration that gives rise to ferroelectric-like responses.
24 Jan 05:39
by Wei Wang, Wenliang Feng, Jun Du, Weinan Xue, Linlin Zhang, Leilei Zhao, Yan Li, Xinhua Zhong
Abstract
The improvement of sunlight utilization is a fundamental approach for the construction of high-efficiency quantum-dot-based solar cells (QDSCs). To boost light harvesting, cosensitized photoanodes are fabricated in this work by a sequential deposition of presynthesized Zn–Cu–In–Se (ZCISe) and CdSe quantum dots (QDs) on mesoporous TiO2 films via the control of the interactions between QDs and TiO2 films using 3-mercaptopropionic acid bifunctional linkers. By the synergistic effect of ZCISe-alloyed QDs with a wide light absorption range and CdSe QDs with a high extinction coefficient, the incident photon-to-electron conversion efficiency is significantly improved over single QD-based QDSCs. It is found that the performance of cosensitized photoanodes can be optimized by adjusting the size of CdSe QDs introduced. In combination with titanium mesh supported mesoporous carbon as a counterelectrode and a modified polysulfide solution as an electrolyte, a champion power conversion efficiency up to 12.75% (Voc = 0.752 V, Jsc = 27.39 mA cm−2, FF = 0.619) is achieved, which is, as far as it is known, the highest efficiency for liquid-junction QD-based solar cells reported.

Co-sensitized photoanodes based on presynthesized quantum dots (QDs) are prepared for the first time by a sequential deposition of Zn–Cu–In–Se and CdSe QDs on mesoporous TiO2 films. With a titanium-mesh-supported mesoporous carbon counter electrode and a polysulfide electrolyte, the conversion efficiency of the co-sensitized cells reaches 12.75% under standard AM 1.5G sunlight illumination, a new record for liquid-junction QD-based cells.