Journal of the American Chemical Society
DOI: 10.1021/jacs.7b12615
The dimetallic system [CuII2(L)]4+ contains two facing equivalent metallocyclam subunits and incorporates ambidentate anions, mono- (halides) and poly-atomic (sulfate), which bridge the two CuII centres. Isothermal titration calorimetry (ITC) experiments in water showed that the log K values of the inclusion equilibria for halides and sulfate varied over a restricted interval (3.6±0.2), which indicated lack of selectivity and that similarity of ΔG° values resulted from the unbalanced contribution of the ΔH° and TΔS° terms: the more favourable the one, the less favourable the other. In particular, a linear dependence of ΔH° and TΔS° was observed (a typical enthalpy/entropy compensatory diagram), which assigned a major role to hydration terms: 1) a more hydrated anion resulted in a more endothermic dehydration process; and 2) a larger number of water molecules released to the solution resulted in a more positive TΔS°. Limiting cases refer to the complexation 1) of the poorly hydrated iodide (highly exothermic process, entropically disfavoured), and 2) of the highly hydrated sulfate (moderately endothermic process, entropically very favoured). Anion receptors operating in water belong to two main domains: 1) those exhibiting positive ΔH° and positive TΔS° (+/+ signature), and 2) those displaying the opposite behaviour: (−/− signature). The receptor investigated herein connects the two domains, along the ΔH°/TΔS° straight line, thanks to the hidden role of the versatile metal–anion interaction.

Selectivity outside the molecule: The dicopper(II) bicyclam receptor [CuII2(L)]4+ includes ambidentate anions (halides, sulfate) in water with poor selectivity (see scheme), which is derived from conflicting enthalpy and entropy contributions: the more favourable the one contribution, the less favourable the other. This indicates the major role of solvational terms (mainly anion dehydration), which mitigate the contribution of the metal–anion interaction.
 Open Access
Open Access   This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.

Following a late-stage functionalization strategy, a series of heteroditopic cryptand receptors were prepared in three steps only from 1,4-dioxane. As evidenced by 1H NMR spectroscopic and solid state analyses, these polyamide–crown ether conjugates present general ion pair binding capacity towards salts of monovalent cations and linear triatomic anions.

A crown in a cage: Using a late-stage functionalization strategy, seven ditopic receptors were prepared in three steps from 1,4-dioxane. 1H NMR spectroscopic and solid state analyses of these novel polyamide–crown ether conjugates present general ion pair binding capacity towards salts made of sodium or potassium cations and linear triatomic anions.
A Saturn-like 1:1 complex composed of macrocyclic oligothiophene E-8T7A and C60 fullerene (C60) was synthesized to investigate the interaction between macrocyclic oligothiophenes and C60 in solution and the solid state. Because the Saturn-like 1:1 complex E-8T7A⋅C60 is mainly stabilized by van der Waals interactions between C60 and the sulfur atoms of the E-8T7A macrocycle, C60 is rather weakly incorporated inside the macro-ring in solution. However, in the solid state the Saturn-like 1:1 complex preferentially formed single crystals or nanostructured polymorphs. Interestingly, X-ray analysis and theoretical calculations exhibited hindered rotation of C60 in the Saturn-like complex due to interactions between C60 and the sulfur atoms. Furthermore, the photoinduced charge transfer (CT) interaction between E-8T7A and C60 in solution was investigated by using femtosecond transient absorption (TA) spectroscopy. The ultrafast TA spectral changes in the photoinduced absorption bands were attributed to the CT process in the Saturn-like structure.

Saturn's rings: The Saturn-like 1:1 complex between C60 and the sulfur atoms of macrocyclic oligothiophene (mainly stabilized by van der Waals interactions) interacts weakly in solution; however, in the solid state this complex preferentially forms single crystals that exhibit hindered rotation of C60 in the complex due to the interactions between C60 and the surrounding sulfur atoms (see figure).
The halogen bond is a supramolecular interaction between a Lewis-acidic region of a covalently bound halogen and a Lewis base. It has been studied widely in silico and experimentally in the solid state; however, solution phase applications have attracted enormous interest in the last years. This Minireview highlights selected recent developments of halogen bond interactions in solution with focus on the use of halogen bond receptors in anion recognition and sensing, anion templated self-assembly as well as in organo-catalysis.
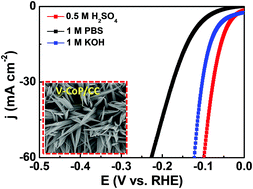
In order to design more powerful electrocatalysts, developing our understanding of the role of the surface structure and composition of widely abundant bulk materials is crucial. This is particularly true in the search for alternative hydrogen evolution reaction (HER) catalysts to replace platinum. We report scanning electrochemical cell microscopy (SECCM) measurements of the (111)-crystal planes of Fe4.5Ni4.5S8, a highly active HER catalyst. In combination with structural characterization methods, we show that this technique can reveal differences in activity arising from even the slightest compositional changes. By probing electrochemical properties at the nanoscale, in conjunction with complementary structural information, novel design principles are revealed for application to rational material synthesis.

Small change, big difference: Local investigation of the hydrogen evolution reaction (HER) on single crystal of iron nickel sulfide, one of the most active non-noble-metal HER catalysts, was carried out using scanning electrochemical cell microscopy (SECCM). Small variations in the Ni/Fe ratio at the surface induce tremendous changes in the catalytic HER activity.
Open Access  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
Water splitting: Passing the acid test
Water splitting: Passing the acid test, Published online: 19 December 2017; doi:10.1038/nchem.2921
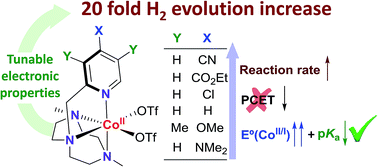
Water-oxidation catalysts that are fast and efficient in strong acid are rare even though there are several benefits for systems working at low pH. Such catalysts usually feature expensive noble metals such as ruthenium and iridum; however, an electrocatalytic system that is exceptionally efficient and based on cobalt has now been developed.

Much like catching a star in the night sky, selective recognition of sulfate anions in an aqueous solution remains a challenge in chemistry. Herein, the design and synthesis of a bright fluorescent sensor for sulfate is reported. Non-covalent interactions that contribute to the formation of receptor–sulfate complex were also studied to understand the sensing mechanism. Inspired by a character from the animated film “Hedgehog in the Fog”, the cover was designed by Alexander Oshchepkov and the authors. More information can be found in the Communication by E. A. Kataev et al. (DOI: 10.1002/chem.201704098).

An electro- and photo-active rotaxane incorporating ten peripheral Zn(II)-porphyrin moieties and a free-base porphyrin stopper has been prepared. Electrochemical measurements revealed that this compound is capable of mimicking the blooming of a flower. On the other hand, steady state investigations have shown that the multi-porphyrinic rotaxane is a light-harvesting device mimicking the antennae of the natural photosynthetic system. More information can be found in the Full Paper by B. Delavaux-Nicot, E. Maisonhaute, J.-F. Nierengarten et al. (DOI: 10.1002/chem.201704124).

The unprecedented application of a chiral halogen-bonding [3]rotaxane host system for the discrimination of stereo- and E/Z geometric isomers of a dicarboxylate anion guest is described. Synthesised by a chloride anion templation strategy, the [3]rotaxane host recognises dicarboxylates through the formation of 1:1 stoichiometric sandwich complexes. This process was analysed by molecular dynamics simulations, which revealed the critical synergy of halogen and hydrogen bonding interactions in anion discrimination. In addition, the centrally located chiral (S)-BINOL motif of the [3]rotaxane axle component facilitates the complexed dicarboxylate species to be sensed via a fluorescence response.

The discrimination between dicarboxylate stereo- and geometric isomers by a chiral halogen-bonding [3]rotaxane can be monitored in terms of the fluorescence response. Computational modelling studies revealed the critical synergy between the axle of the rotaxane host and macrocycle components in achieving dicarboxylate guest selectivity.
Borinic acids have typically not been considered as hydrogen bond donor groups in molecular recognition. Described herein is a bifunctional borane/borinic acid derivative (2) in which the two functionalities are connected by a 1,8-biphenylenediyl backbone. Anion binding studies reveal that 2 readily binds a fluoride anion by formation of a unique B−F⋅⋅⋅H−O−B hydrogen bond. This hydrogen bond is characterized by a short H-F distance of 1.79(3) Å and a large coupling constant (1JHF) of 57.2 Hz. The magnitude of this interaction, which has also been investigated computationally, augments the fluoride anion binding properties of 2, thus making it compatible with aqueous environments.

A Brønsted and Lewis handshake: The borinic acid functionality of a new diboron electrophilic host reaches across the binding pocket to engage a borane-bound fluoride anion in a strong hydrogen-bonding interaction. The resulting B−F⋅⋅⋅H−O−B interaction stabilizes the complex in aqueous solutions, thereby illustrating the role that borinic acids may play as hydrogen bond donor groups.
Metallic-phase molybdenum disulfide (1T-MoS2) nanosheets have proven to be highly active in the hydrogen evolution reaction (HER). We describe construction of photosensitizer functionalized 1T-MoS2 by covalently tethering the molecular photosensitizer [RuII(bpy)3]2+ (bpy=2,2′-bipyridine) on 1T-MoS2 nanosheets. This was achieved by covalently tethering the bpy ligand to 1T-MoS2 nanosheets, and subsequent complexation with [RuII(bpy)2Cl2] to yield [RuII(bpy)3]–MoS2. The obtained [RuII(bpy)3]–MoS2 nanosheets were characterized using infra-red, electronic absorption, X-ray photoelectron, and Raman spectroscopies, X-ray powder diffraction and electron microscopy. The fabricated material exhibited a significant improvement of photocurrent and HER performance, demonstrating the potential of such two-dimensional [RuII(bpy)3]–MoS2 constructs in photosensitized HER.

Catalyzing evolution: Covalent tethering of 1T-MoS2 nanosheets with [RuII(bpy)3]2+ photosensitizers yields photosensitizing H2-evolution catalysts. The [RuII(bpy)3]–MoS2 hybrid exhibited a significantly enhanced photocurrent and H2-evolution performance.
In the field of renewable energy, the splitting of water into hydrogen and oxygen fuel gases using water electrolysis is a prominent topic. Traditionally, these catalytic processes have been performed by platinum-group metal catalysts, which are effective at promoting water electrolysis but expensive and rare. The search for an inexpensive and Earth-abundant catalyst has led to the development of 3d-transition-metal phosphides for the hydrogen evolution reaction. These catalysts have shown excellent activity and stability. In this review, we discuss the electronic and crystal structures of bulk and surface of selected Fe, Co, and Ni phosphides, and their relationships to the experimental catalytic activity. The various synthetic protocols towards the state-of-the-art transition metal phosphide electrocatalysts are also discussed.

Transition-metal phosphides, which accommodate a number of stoichiometries, peculiar crystal arrangements, and diverse electronic structures, are emerging as the highly active and stable electrocatalysts for water reduction. In this Minireview, the relationship between the structure and the resultant activity is explored, and can be used as a useful concept for the replacement of Pt in the cathode for water electrolysis.
Artificial molecular motors hold great promise for application in responsive functional materials as well as to control the properties of biohybrid systems. Herein a strategy is reported to modulate the rotation of light-driven molecular motors. That is, the rotary speed of a molecular motor, functionalized with a biphenol moiety, could be decreased in situ by non-covalent substrate binding, as was established by 1H NMR and UV/Vis spectroscopy. These findings constitute an important step in the development of multi-responsive molecular machinery.

The rotation of a light-driven molecular motor can be controlled reversibly by supramolecular binding of a diamine guest to a biphenol moiety.
π-Conjugated molecular cages are very challenging targets in structural organic chemistry, supramolecular chemistry, and materials science. The synthesis and physical characterizations are reported of the first three-dimensionally π-conjugated diradical molecular cage PTM-C, in which two polychlorotriphenylmethyl (PTM) radicals are linked by three bis(3,6-carbazolyl) bridges. This cage compound was synthesized mainly by intermolecular Yamamoto coupling followed by deprotonation and oxidation. It is stable and its structure was confirmed by X-ray crystallographic analysis. The two carbon-centered PTM radicals are weakly coupled through electronic interactions with the carbazole spacers, as revealed by optical, electronic, and magnetic measurements as well as theoretical calculations.

3D diradical cage! A diradical molecular cage that is π-conjugated in three dimensions was synthesized, and the cage structure was confirmed by X-ray crystallographic analysis. 3D π-conjugation and weak intramolecular spin exchange interaction were verified by optical, electronic, and magnetic measurements, assisted by DFT calculations.