Nature Nanotechnology. doi:10.1038/nnano.2016.222
Author: Ayusman Sen
Nanostructured photocatalytic microrods can swim either towards or away from a light source depending on their surface functionalization.
Nature Nanotechnology. doi:10.1038/nnano.2016.222
Author: Ayusman Sen
Nanostructured photocatalytic microrods can swim either towards or away from a light source depending on their surface functionalization.
Organic solar cells (OSCs) are lightweight, have adaptable colors, and can be produced in low-cost procedures on transparent and flexible surfaces. This makes them attractive for markets in which other technologies cannot compete, for example in architectural and consumer product integration. However, both efficiencies and long term operational stability of OSCs do not yet meet the standards set by their inorganic counterparts. This review compiles the growing knowledge about how nanostructured carbon materials, such as fullerenes and carbon nanotubes, decisively influence the operational stability of organic photovoltaics. Firstly, important degradation pathways are introduced and a differential detection scheme is set up to find the dominant loss channel by means of state-of-the-art characterization methods. Then, fullerenes ability to both stabilize and destabilize the donor polymer against photooxidation via different mechanisms (e.g., inner filter effect or radical scavenging) is examined in detail. The “burn-in” problem, an initial rapid efficiency loss in PC60BM-based OSCs, is shown to derive from light-induced PC60BM dimerization, an effect that can also be positively exploited to reduce thermal degradation. Finally, thermal stabilization via additional approaches involving the fullerene derivative, such as crosslinking or incorporation into block copolymers, is presented.

Carbon nanostructures, particularly fullerene derivatives, possess the ability to act as radical scavengers and undergo photodimerization, consequently influencing the photochemical and morphological degradation of the active layer. These properties can be exploited to produce organic solar cells with longer operational lifetimes without compromising device efficiency and manufacturing costs.


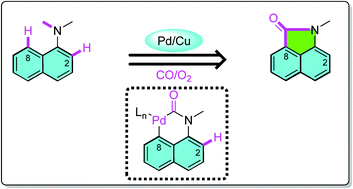
A novel reagent, which introduces two sulfur atoms in one step, was designed and used for the construction of diverse disulfanes by copper-catalyzed oxidative cross-coupling under mild reaction conditions. By applying this stable and readily prepared reagent, late-stage modification of pharmaceuticals and natural products can be achieved straightforward. The scaled-up experiments further indicated the practicality of this protocol. The pH value of the system plays a key role in achieving highly selective cleavage of the C−S bond instead of a S−S bond in the transformation.

Double S: A novel reagent, which introduces two sulfur atoms in one step, was designed for the construction of various disulfanes by copper-catalyzed oxidative cross-coupling under mild reaction conditions. This reagent can be used for late-stage modification of pharmaceuticals and natural products. The pH value of system plays a key role in the selective cleavage of a C−S bond instead of a S−S bond. Boc=tert-butoxycarbonyl.
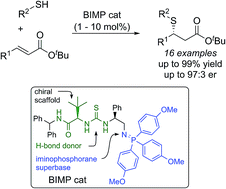
UV-induced disulfide formation (UV-DF) and disulfide reduction (UV-DR) reactions for surface functionalization and dynamic photopatterning are presented. Both photochemical reactions allow for the spatially and temporally controlled, reversible transition between thiol- and disulfide-functionalized surfaces. The dynamic photopatterning strategy was demonstrated by the UV-induced attachment, exchange, and detachment on thiol-modified substrates.

Shine a light on thiols: UV-induced disulfide formation (UV-DF) and disulfide reduction (UV-DR) reactions are demonstrated to be effective for reversible surface modification, patterning, and attachment and detachment of functional groups. This photodynamic thiol-disulfide exchange process will be useful for the design of novel dynamic and responsive functional interfaces and micropatterns.


Marcin A. Majewski, Yongseok Hong, Prof. Tadeusz Lis, Dr. Janusz Gregoliński, Prof. Piotr J. Chmielewski, Dr. Joanna Cybińska, Prof. Dongho Kim and Prof. Marcin Stępień

A non-Euclidean belt: Octulene, the largest coronoid ring reported to date, features a saddle-shaped aromatic hydrocarbon ring that fits on a hyperbolic surface. The large central cavity, in spite of its low electrostatic potential, acts as an anion receptor in nonpolar media, with a particularly good affinity for the chloride anion.
Nature Nanotechnology. doi:10.1038/nnano.2016.169
Authors: Charalampos G. Pappas, Ramim Shafi, Ivan R. Sasselli, Henry Siccardi, Tong Wang, Vishal Narang, Rinat Abzalimov, Nadeesha Wijerathne & Rein V. Ulijn


A covalent organic helical cage (COHC) with D3 symmetry bearing two 1,3,5-trimethylphenyl cores and six di-tert-butyldiethynylallene moieties was synthesized and fully characterized. This molecular structure cage, unlike a previously reported one, favors inclusion-complex formation with organometallic sandwich compounds due to the presence of methyl groups on the aryl rings. The strong chiroptical responses of these COHCs, along with their ability to entrap guest molecules, enabled the detection of a ruthenium sandwich compound by means of electronic circular dichroism (ECD) spectroscopy.

Covalent organic helical cages (COHCs) are valuable containers for organometallic sandwich compounds. The ruthenium inclusion complex can be chiroptically detected.
We report two new helicenes derived from the double fusion of an acene with two perylene diimide (PDI) subunits. These PDI-helicene homologs exhibit very different structural and electronic properties, despite differing by only a single ring in the link between the PDI units. The shorter inter-PDI link brings the two PDI subunits closer together, and this results in the collision of their respective π-electron clouds. This collision facilitates intramolecular through-space electronic delocalization when the PDI-helicene is reduced.

Two helicenes were synthesized by double fusion of an acene with two perylene diimide (PDI) subunits. These PDI-helicene homologs show different structural and electronic properties, despite differing by only a single ring in the link between the PDI units. The shorter link brings the two PDI subunits closer together, and this results in the collision of their respective π-electron clouds.


Topological molecular connections and structures, including physical entanglements in polymer networks, knots along polymer chains, and rotaxanes in sliding ring gels, have important consequences for the physical properties of polymeric materials. Often these topologies contribute through their ability to bear mechanical stress, but experimental measures of their relative mechanical strength are rare. Here, we use sonochemical polymer mechanochemistry to assess the relative mechanical strength of a multicatenane copolymer relative to copolymers of cyclic and linear analogs. The relative mechanical strengths are obtained by comparing the limiting molecular weights (Mlim) and contour lengths (Llim) of the polymers under pulsed ultrasound of their dilute solutions. The values of Mlim and Llim, and thus the inferred mechanical strengths of the polymers, are effectively identical. The mechanical bonds of the catenanes are therefore as strong, or stronger, mechanically as the covalent bonds along the polymer backbone.

Polymer chain scission by sonication: The relative mechanical strength of poly([2]catenane) copolymers and copolymers of cyclic and linear analogs was studied. Sonochemical polymer mechanochemistry of poly([2]catenane) copolymers showed that the mechanical strength of the mechanical bond is as strong or stronger than that of a conventional linear polymer backbone.
 Open Access
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.


Organic solar cells hold promise of providing low-cost, renewable power generation, with current devices providing up to 13% power conversion efficiency. The rational design of more performant systems requires an in-depth understanding of the interactions between the electron donating and electron accepting materials within the active layers of these devices. Here, we explore works that give insight into the intermolecular interactions between electron donors and electron acceptors, and the impact of molecular orientations and environment on these interactions. We highlight, from a theoretical standpoint, the effects of intermolecular interactions on the stability of charge carriers at the donor/acceptor interface and in the bulk and how these interactions influence the nature of the charge transfer states as well as the charge separation and charge transport processes.

An assessment of intermolecular interactions and their impact on electronic processes in organic solar cells is presented. While a great deal has been learned about the molecular-scale optical and electronic processes in these devices, a complete understanding of how the active-layer composition and morphology influence the charge transfer states, polarization and charge separation still needs to be reached.
Light effect on Click reaction: Role of photonic quantum dot catalyst
Scientific Reports, Published online: 13 September 2016; doi:10.1038/srep33025
Since the advent of mechanically interlocked molecules (MIMs), many approaches to templating their formation using various different noncovalent bonding interactions have been introduced and explored. In particular, employing radical-pairing interactions between BIPY.+ units, the radical cationic state of 4,4′-bipyridinium (BIPY2+) units, in syntheses is not only a convenient but also an attractive source of templation because of the unique properties residing in the resulting catenanes and rotaxanes. Herein, we report a copper-mediated procedure that enables the generation, in the MIM-precursors, of BIPY.+ radical cations, while the metal itself, which is oxidized to CuI, catalyzes the azide–alkyne cycloaddition reactions that result in the efficient syntheses of two catenanes and one rotaxane, assisted by radical-pairing interactions between the BIPY.+ radical cations. This procedure not only provides a fillip for making and investigating the properties of Coulombically challenged catenanes and rotaxanes, but it also opens up the possibility of synthesizing artificial molecular machines which operate away from equilibrium.

A user-friendly technique for the construction of positively charged catenanes and rotaxanes, templated by radical-pairing interactions in a convenient and efficient manner, has been conceived and implemented. This method opens up the possibility of producing integrated systems with Coulombically challenged catenanes and rotaxanes and of fabricating devices based on these systems.