Inorganic Chemistry
DOI: 10.1021/acs.inorgchem.6b02991
 Open Access
Open Access   This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.


Open Access   This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.



The reaction of the 2-(trimethylsilyl)imidazolium triflate 9 with diarylboron halides (4-R-C6H4)2BX (R=H, X=Br; R=CH3, X=Cl; R=CF3, X=Cl) afforded the NHC-stabilized borenium cations 10 a–c. Cyclic voltammetry revealed a linear correlation between the Hammett parameter σp of the para substituent and the half-wave potential. Chemical reduction with decamethylcobaltocene, [(C5Me5)2Co], furnished the corresponding radicals 11 a–c; their characterization by EPR spectroscopy confirmed the paramagnetic character of 11 a–c, with large hyperfine coupling constants to the boron isotopes 11B and 10B, while delocalization of the unpaired electron into the NHC is negligible. DFT calculations of the percentage of spin density distribution between the carbene (NHC) and the boryl fragments (BR2) revealed for 11 a–c a spin density ratio (BR2/NHC) of ca. 9:1, which underlines their distinct boryl radical character. The molecular structure of the most stable species 11 c was established by X-ray diffraction analysis.

Extended expiration date: The reaction between a frustrated N-heterocyclic carbene/silylium ion Lewis pair and diarylboron halides afforded a series of NHC-supported borenium cations. One-electron reduction gave persistent boryl radicals that were studied by EPR spectroscopy, X-ray diffraction analysis, and DFT methods, revealing almost exclusive localization of the unpaired electron on the diarylboryl fragments.
The neutral radical (Me2-cAAC)2AlCl2 (2) is stabilized by cyclic (alkyl)(amino)carbenes (cAACs). Complex 2 was synthesized by reduction of the Me2-cAAC:![[RIGHTWARDS ARROW]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/002192.gif) AlCl3 (1) adduct with KC8 in the presence of another equivalent of Me2-cAAC. The crystal structure of 2 shows that the Al−C bond lengths of the two carbenes bound to the Al center are considerably different, which is likely the result of intermolecular interactions. Quantum-chemical calculations from the gas phase give an equilibrium structure with identical Al−C bond lengths. Compound 2 exhibits monoradical character, which was confirmed by EPR measurements. A bonding analysis indicates that the unpaired electron resides mainly at the carbene carbon atoms. Compound 2 is an example for an unusual neutral Al radical.
AlCl3 (1) adduct with KC8 in the presence of another equivalent of Me2-cAAC. The crystal structure of 2 shows that the Al−C bond lengths of the two carbenes bound to the Al center are considerably different, which is likely the result of intermolecular interactions. Quantum-chemical calculations from the gas phase give an equilibrium structure with identical Al−C bond lengths. Compound 2 exhibits monoradical character, which was confirmed by EPR measurements. A bonding analysis indicates that the unpaired electron resides mainly at the carbene carbon atoms. Compound 2 is an example for an unusual neutral Al radical.

A monomeric aluminum radical was synthesized from its cyclic (alkyl)(amino)carbene adduct, cAACAlCl3, by reduction with KC8. The AlCl2 center is stabilized by two cAACs and shows monoradical character. EPR spectroscopy and theoretical calculations provided further insight into the properties of the radical species and its resonance structures.

Reaction of the donor-stabilized silylene [iPrNC(NiPr2)NiPr]2Si (1) with FeBr2, CoBr2, NiBr2⋅MeOCH2CH2OMe, ZnCl2, and ZnBr2 afforded the respective transition-metal silylene complexes 4–8, the formation of which can be described in terms of a Lewis acid/base reaction (4, 5, 7, 8) or a nucleophilic substitution reaction (6). However, the reactivity profile of silylene 1 is not only based on its strong Lewis base character; the different coordination modes of the two guanidinato ligands (4–6 vs. 7 and 8) add an additional reactivity facet. The paramagnetic compounds 4 and 5 and the diamagnetic compounds 6⋅THF, 7, and 8⋅0.5 Et2O were structurally characterized by single-crystal X-ray diffraction. In addition, compound 6⋅THF was studied by 15N and 29Si solid-state NMR spectroscopy, and 7 and 8 were characterized by NMR spectroscopic studies in the solid state (15N, 29Si) and in solution (1H, 13C, 29Si). Compounds 4–8 represent very rare examples of FeII, CoII, NiII, and ZnII silylene complexes. Four-coordinate silicon(II) compounds with an SiN3M skeleton (M=Fe, Co, Ni) and M in the formal oxidation state +2 (4–6) have not yet been reported, and five-coordinate silicon(II) compounds with an SiN4Zn skeleton (7, 8) are also unprecedented.

Transition-metal silylene complexes: The donor-stabilized silylene [iPrNC(NiPr2)NiPr]2Si reacts with FeBr2, CoBr2, NiBr2⋅MeOCH2CH2OMe, ZnCl2, and ZnBr2 to afford compounds 1–5, a series of very rare examples of FeII, CoII, NiII, and ZnII silylene complexes. The reactivity profile of the silylene is not only based on its strong Lewis base character; the different coordination modes of the two guanidinato ligands (bridging vs. nonbridging) add an additional reactivity facet.
An aluminum-catalyzed hydroboration of alkynes using either the commercially available aluminum hydride DIBAL-H or bench-stable Et3Al⋅DABCO as the catalyst and H-Bpin as both the boron reagent and stoichiometric hydride source has been developed. Mechanistic studies revealed a unique mode of reactivity in which the reaction is proposed to proceed through hydroalumination and σ-bond metathesis between the resultant alkenyl aluminum species and HBpin, which acts to drive turnover of the catalytic cycle.

Main-group catalysts: An aluminum-catalyzed hydroboration of alkynes using either the commercially available aluminum hydride DIBAL-H or bench-stable Et3Al⋅DABCO as the catalyst and H-Bpin as both the boron reagent and stoichiometric hydride source has been developed. Mechanistic studies revealed a unique mode of reactivity, in which the reaction is proposed to proceed through hydroalumination and σ-bond metathesis between the resultant alkenyl aluminum species and HBpin.

We report on the synthesis and structural characterization of unprecedented anionic parent compounds of mixed Group 13/15 elements. The reactions of the pnictogenylboranes H2E-BH2⋅NMe3 (1 a=P, 1 b=As) with phosphorus and arsenic centered nucleophiles of the type [EH2]− (E=P, As) lead to the formation of compounds of the type [H2E-BH2-E′H2]− (2: E=E′=P; 3: E=E′=As; 4: E=P, E′=As) containing anionic pnictogen–boron chain-like units. Furthermore, a longer 5-membered chain species [H2As-BH2-PH2-BH2-AsH2]− (5) and a cyclic compound [NHCdipp-H2B-PH2-BH2-NHCdipp]+[P5B5H19]− (6) containing a n-butylcyclohexane-like anion were obtained. All the compounds have been characterized by X-ray structure analysis, multinuclear NMR spectroscopy, IR spectroscopy, and mass spectrometry. DFT calculations elucidate their high thermodynamic stability, the charge distribution, and give insight into the reaction pathway.

Finally, thoroughly negative! Using alkali-metal salts of phosphanide and arsenide, the parent compounds of the phosphanyl- and arsanylboranes can be easily converted into linear anionic chain molecules. They are unique representatives of the anionic class of pnictogenylboranes and could be structurally characterized.
Phosphine-stabilized silylenes react with silanes and a phosphine by silylene insertion into E−H σ-bonds (E=Si,P) at room temperature to give the corresponding silanes. Of special interest, the process occurs reversibly at room temperature. These results demonstrate that both the oxidative addition (typical reaction for transient silylenes) and the reductive elimination processes can proceed at the silicon center under mild reaction conditions. DFT calculations provide insight into the importance of the coordination of the silicon center to achieve the reductive elimination step.

New inserts: Phosphine-stabilized silylenes react with silanes (or phosphine) by silylene insertion into E−H σ-bonds (E=Si,P) at room temperature to give the corresponding silanes. Of special interest, the process occurs reversibly at room temperature. These results demonstrate that both the oxidative addition and the reductive elimination processes can proceed at the silicon center under mild reaction conditions.
Stabilized borylenes (L2BH:) with weakly π-accepting substituents L, such as phosphines, were previously believed to be unstable. In the current manuscript, we describe a series of complexes formally containing a phosphine-stabilized borylene or boryl anion. In contrast to common trivalent boron compounds, the boron-based ligands in this study act as electron-donating ligands. The reported iron hydride complexes exhibit a unique reactivity pattern, undergoing a reversible B−H reductive elimination concomitant with oxidation of the boron(I) center.

As a free molecule, a phosphine-stabilized borylene is unstable, but it was isolated as an electron-donating ligand in an iron complex. This complex exhibits a unique reactivity pattern, undergoing a reversible B−H reductive elimination concomitant with oxidation of the boron(I) center.


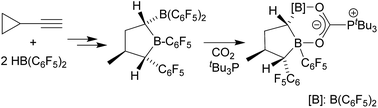
We report a new class of frustrated Lewis pairs (FLPs) by the hydroboration of bulky isocyanates iPr2ArNCO (iPr2Ar=2,6-iPr2C6H3) and Ph2tBuArNCO (Ph2tBuAr=2,6-Ph2-4-tBuC6H2) with Piers’ borane (HB(C6F5)2). While hydroboration of smaller isocyanates such as iPr2ArNCO leads to isocyanate—N/B FLP adducts, hydroboration of the bulkier Ph2tBuArNCO allows isolation of the substrate-free aminoborane with a short, covalent N−B bond. This confused FLP reversibly binds unsaturated substrates such as isocyanates and isocyanides, suggesting the intermediacy of a “normal” FLP along the reaction pathway, supported by high-level DFT studies and variable-temperature NMR spectroscopy. These results underscore the possibility of FLP behavior in systems that possess no obvious frustrated Lewis acid–base interaction.

A confused isomer: A new class of frustrated Lewis pairs (FLPs) resulted from hydroboration of isocyanates, which provided N-formyl aminoboranes (see picture). This “confused” isomer showed the typical behavior of a frustrated Lewis pair through interconversion with its frustrated form, such as capture of unsaturated substrates.
Reaction of a N-heterocyclic silylene (NHSi) with PhBX2 (X=Cl, Br) readily afforded six-membered silaborinines through an insertion/ring expansion sequence. Increasing the sterics of the borane from phenyl to duryl enabled the selective generation and isolation of the highly colored silylborane intermediates. Theoretical studies on the mechanism and energetics of the silaborinine formation were fully consistent with the experimental observations.

Size matters: Reaction of an N-heterocyclic silylene with aryl-substituted dihaloboranes ArBX2 (Ar=Ph, Dur; X=Cl, Br) readily afforded either silaborinines (Ar=Ph; insertion/ring expansion) or silylborane intermediates (Ar=Dur; insertion). Dur=2,3,5,6-tetramethylphenyl.

Reduction of carbene-borane adduct [(cAAC)BBr2(CN)] (cAAC=1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene) cleanly yielded the tetra(cyanoborylene) species [(cAAC)B(CN)]4 presenting a 12-membered (BCN)4 ring. The analysis of the Kohn–Sham molecular orbitals showed significant borylene character of the BI atoms. [(cAAC)B(CN)]4 was found to reduce two equivalents of AgCN per boron center to yield [(cAAC)B(CN)3] and fragmented into two-coordinate boron(I) units upon reaction with IMeMe (1,3,4,5-tetramethylimidazol-2-ylidene) to yield the corresponding tricoordinate mixed cAAC-NHC cyanoborylene. The analogous cAAC-phosphine cyanoborylene was obtained by reduction of [(cAAC)BBr2(CN)] in the presence of excess phosphine.

All square: Reduction of a carbene-borane adduct provides the tetra(cyanoborylene) species [(cAAC)B(CN)]4 with a 12-membered (BCN)4 ring. This species was found to reduce two equivalents of AgCN to yield [(cAAC)B(CN)3] and fragmented into two-coordinate boron(I) units upon reaction with an NHC.

Hexachlorodisilane reacts with diarylmethanones (aryl=C6H5, 4-MeC6H4, 4-tBuC6H4, 4-ClC6H4, 4-BrC6H4) to furnish the corresponding tetraarylethylenes in good yields. The reductive conversion requires temperatures of about 160 °C and reaction times of 60–72 h. In the initial step, the Lewis-basic carbonyl groups likely generate low-valent [SiCl2] as an analogue of [TiCl2] in the classical McMurry reaction. The coupling sequence further proceeds via benzopinacolones, which have been isolated as key intermediates.

Successful with silanes: Diarylmethanones Ar2C=O react with Si2Cl6 at 160 °C to give tetraarylethylenes in good yields (see scheme). This novel reductive coupling reaction likely proceeds via intermediary [SiCl2] generation and is thus related to the [TiCl2]-mediated McMurry reaction.
A new approach to main-group H2 activation combining concepts of transition-metal and frustrated Lewis pair chemistry is reported. Ambiphilic, metal-like reactivity toward H2 can be conferred to 9,10-dihydro-9,10-diboraanthracene (DBA) acceptors by the injection of two electrons. The resulting [DBA]2− ions cleave the H−H bond with the formation of hydridoborates under moderate conditions (T=50–100 °C; p<1 atm). Depending on the boron-bonded substituents R, the addition is either reversible (R=C≡CtBu) or irreversible (R=H). The reaction rate is strongly influenced by the nature and the coordination behavior of the countercation (Li+ slower than K+). Quantum-chemical calculations support the experimental observations and suggest a concerted, homolytic addition of H2 across both boron atoms. As proven by the successful conversion of Me3SiCl into Me3SiH, the system Li2[DBA]/H2 appears generally relevant for the hydrogenation of element–halide bonds.

Borane metamorphosis: The injection of two electrons transformed the ditopic Lewis acid diboraanthracene into a main-group ambiphile (MGA) that activated H2 much as a transition metal would. The H2 molecule underwent selective addition across the two boron atoms, and the resulting hydridoborate could be used as a hydride-transfer reagent.