16 Feb 11:41
Green Chem., 2021, 23,2198-2232
DOI: 10.1039/D0GC04430J, Critical Review
Emmanuel Isaac Akpan, Bernd Wetzel, Klaus Friedrich
Eco-friendly alternative processes are required for functionally modified wood to maintain low embodied energy and near zero emission levels.
The content of this RSS Feed (c) The Royal Society of Chemistry

13 Feb 09:27
by Ruslan Muhamadejev, Renate Melngaile, Paula Paegle, Ieva Zibarte, Marina Petrova, Kristaps Jaudzems, and Janis Veliks
![TOC Graphic]()
The Journal of Organic Chemistry
DOI: 10.1021/acs.joc.0c02744

12 Feb 21:47
by Sean M. Treacy and Tomislav Rovis
![TOC Graphic]()
Journal of the American Chemical Society
DOI: 10.1021/jacs.1c00687

12 Feb 13:55
Chem. Sci., 2021, 12,4237-4266
DOI: 10.1039/D0SC06000C, Review Article

Open Access
Margery Cortes-Clerget, Julie Yu, Joseph R. A. Kincaid, Peter Walde, Fabrice Gallou, Bruce H. Lipshutz
A review that highlights water as the logical reaction medium in which organic chemistry can be practiced. The key roles that water can play in directing reaction outcomes, including impacting mechanistic features, are discussed using selected examples.
The content of this RSS Feed (c) The Royal Society of Chemistry

12 Feb 13:51
by Yan Wan and Jong-Min Lee
![TOC Graphic]()
ACS Catalysis
DOI: 10.1021/acscatal.0c05419

12 Feb 13:51
by Jun-Jie Tian, Ning Liu, Qi-Fei Liu, Wei Sun, and Xiao-Chen Wang

Journal of the American Chemical Society
DOI: 10.1021/jacs.1c00006

12 Feb 13:48
by Featherston, A. L., Kwon, Y., Pompeo, M. M., Engl, O. D., Leahy, D. K., Miller, S. J.
We report the catalytic stereocontrolled synthesis of dinucleotides. We have demonstrated, for the first time to our knowledge, that chiral phosphoric acid (CPA) catalysts control the formation of stereogenic phosphorous centers during phosphoramidite transfer. Unprecedented levels of diastereodivergence have also been demonstrated, enabling access to either phosphite diastereomer. Two different CPA scaffolds have proven to be essential for achieving stereodivergence: peptide-embedded phosphothreonine-derived CPAs, which reinforce and amplify the inherent substrate preference, and C2-symmetric BINOL-derived CPAs, which completely overturn this stereochemical preference. The presently reported catalytic method does not require stoichiometric activators or chiral auxiliaries and enables asymmetric catalysis with readily available phosphoramidites. The method was applied to the stereocontrolled synthesis of diastereomeric dinucleotides as well as cyclic dinucleotides, which are of broad interest in immuno-oncology as agonists of the stimulator of interferon genes (STING) pathway.
11 Feb 14:36
by Dace Cīrule, Irina Novosjolova, Ērika Bizdēna and Māris Turks
Abstract
A new approach was designed for the synthesis of C6-substituted 2-triazolylpurine derivatives. A series of substituted products was obtained in SNAr reactions between 2,6-bistriazolylpurine derivatives and O- and C-nucleophiles under mild conditions. The products were isolated in yields up to 87%. The developed C–O and C–C bond forming reactions clearly show the ability of the 1,2,3-triazolyl ring at the C6 position of purine to act as leaving group.
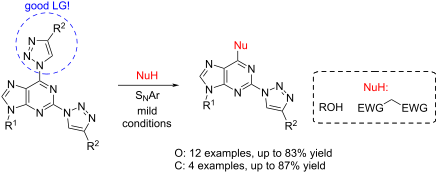
Beilstein J. Org. Chem. 2021, 17, 410–419. doi:10.3762/bjoc.17.37
11 Feb 11:55
by Michelina Soccio, Rita Mazzoni, Carlo Lucarelli, Silvia Quattrosoldi, Andrea Cingolani, Maurizio Fiorini, Nadia Lotti, and Tommaso Tabanelli

ACS Sustainable Chemistry & Engineering
DOI: 10.1021/acssuschemeng.0c05177
11 Feb 09:34
by Thomas Shilpa,
Mohan Neetha,
Gopinathan Anilkumar

Abstract
Recalling the serious hazards imposed by the “ungreen” techniques used over the years, chemists are now expected to adopt synthetic tactics that strictly adhere to the three E's of green chemistry: Environmental, Economical and Energy efficient. Among the various measures undertaken to improve the benignity of a chemical system, top priority has been given for identifying abundant, non‐toxic and non‐flammable greener solvents such as water, replacing detrimental organic solvents. Despite the safer aspects, reactions in aqueous solutions hardly meet the green chemical standards, particularly due to the challenging product isolation and purification techniques. Sharpless's demonstration of, until then almost unknown, “on water” effect was a major breakthrough in the realm of sustainable organic chemistry. Needless to say, integration of homo‐/heterogeneous water‐compatible catalysts under “on water” conditions without deteriorating its efficiency, has also awakened the industrial prospects of the protocol. Among the various transition metals, copper complexes or salts are considered as first line catalysts due to their irrevocable contributions to the synthetic world. Also, due to the water tuneable nature of these catalysts, many copper‐catalysed organic transformations that once required harsh conditions, could now be obtained using clean and safe protocols. This review comprehends the recent advancements in the copper‐catalysed “on water” reactions covering literatures since 2006.
11 Feb 09:32
by Xiao-Feng Wu,
Tim Meyer,
Jabor Rabeah,
Angelika Brückner

The first example on oxidative carbonylation of amines to oxalamides driven by visible light with the release of hydrogen gas has been developed. The new approach uses a simple robust Pd complex, which can even be partially recycled. A mechanistic reason is provided and supported by control experiments and EPR studies, showing that PdI formed and Pd0 was the active species. Both nitrogen‐ and the intermediate acyl radical were detected.
Abstract
The palladium‐catalyzed oxidative carbonylation of amines toward the synthesis of oxalamides has been established around 30 years ago and it usually needs the presence of (over)stoichiometric amounts of oxidant. In this work, the first transformation of this type in which the oxidant was replaced by visible light is described. The new approach uses a simple robust Pd complex, which can even be partially recycled. A mechanistic reason is provided and supported by control experiments and EPR studies, showing that PdI was formed and Pd0 was the active species. Both nitrogen‐ and the intermediate acyl radical can be detected. Moreover, the formation of hydrogen was confirmed by gas GC.
11 Feb 09:32
by You‐Quan Zou,
Niklas Wolff,
Michael Rauch,
Moran Feller,
Quan‐Quan Zhou,
Aviel Anaby,
Yael Diskin‐Posner,
Linda J. W. Shimon,
Liat Avram,
Yehoshoa Ben‐David,
David Milstein

A homogeneous ruthenium‐catalyzed reforming of aqueous ethylene glycol to glycolic acid and hydrogen is described. A plausible reaction mechanism, involving metal–ligand cooperation is proposed and supported by stoichiometric reactions, NMR studies, X‐ray crystallography and computational studies.
Abstract
Glycolic acid is a useful and important α‐hydroxy acid that has broad applications. Herein, the homogeneous ruthenium catalyzed reforming of aqueous ethylene glycol to generate glycolic acid as well as pure hydrogen gas, without concomitant CO2 emission, is reported. This approach provides a clean and sustainable direction to glycolic acid and hydrogen, based on inexpensive, readily available, and renewable ethylene glycol using 0.5 mol % of catalyst. In‐depth mechanistic experimental and computational studies highlight key aspects of the PNNH‐ligand framework involved in this transformation.
10 Feb 17:41
by Ria M. Abdilla-Santes, Jozef G.M. Winkelman, Ilona van Zandvoort, Bert M. Weckhuysen, Pieter C.A. Bruijnincx, Edita Jurak, Peter J. Deuss, and Hero J. Heeres
![TOC Graphic]()
ACS Sustainable Chemistry & Engineering
DOI: 10.1021/acssuschemeng.0c06579
10 Feb 17:39
by Xu-Long Qin, Ang Li, and Fu-She Han

Journal of the American Chemical Society
DOI: 10.1021/jacs.1c00277

10 Feb 17:13
Chem. Sci., 2021, 12,3857-3870
DOI: 10.1039/D0SC06937J, Review Article
Open Access
Trisha Bhattacharya, Animesh Ghosh, Debabrata Maiti
Among numerous solvents available for chemical transformations, 1,1,1,3,3,3-hexafluoro-2-propanol (popularly known as HFIP) has attracted enough attention of the scientific community in recent years.
The content of this RSS Feed (c) The Royal Society of Chemistry

10 Feb 17:06
by Christopher J. J. Hall,
William R. F. Goundry,
Timothy James Donohoe

The scope of carbon–carbon bond‐forming hydrogen‐borrowing reactions has been expanded to include 1,2‐amino alcohols derived from both natural and unnatural amino acids. The vulnerable amine stereocentre is preserved in the reaction by the use of a bulky nitrogen protecting group (trityl or benzyl), with the products being readily cleaved under acidic conditions to the corresponding γ‐aminobutyric acids without further purification.
Abstract
For the first time we have been able to employ enantiopure 1,2‐amino alcohols derived from abundant amino acids in C−C bond‐forming hydrogen‐borrowing alkylation reactions. These reactions are facilitated by the use of the aryl ketone Ph*COMe. Racemisation of the amine stereocentre during alkylation can be prevented by the use of sub‐stoichiometric base and protection of the nitrogen with a sterically hindered triphenylmethane (trityl) or benzyl group. The Ph* and trityl groups are readily cleaved in one pot to give γ‐aminobutyric acid (GABA) products as their HCl salts without further purification. Both steps may be performed in sequence without isolation of the hydrogen‐borrowing intermediate, removing the need for column chromatography.
10 Feb 13:48
by Flora Graham
Nature, Published online: 09 February 2021; doi:10.1038/d41586-021-00374-8
Collaborative online games to help with social-distancing blues. Plus, hundreds of ‘predatory’ journals indexed on Scopus and a call to invest now in variant-proof vaccines.

10 Feb 11:07
Green Chem., 2021, 23,1495-1535
DOI: 10.1039/D0GC03982A, Critical Review
Sami Fadlallah, Pallabi Sinha Roy, Gil Garnier, Kei Saito, Florent Allais
The green aspects of the lignin-derived monomers and polymers have been analysed. A different viewpoint has been provided to encourage researchers to use simple and yet effective green metrics calculations in the development of sustainable syntheses.
The content of this RSS Feed (c) The Royal Society of Chemistry

10 Feb 09:22
Chem. Commun., 2021, 57,3026-3029
DOI: 10.1039/D1CC00181G, Communication
Akash Jana, Amol Kumar, Biplab Maji
C-Alkylations of nine different classes of methyl-substituted N-heteroarenes are disclosed using a bench stable Mn(I)-catalyst under borrowing hydrogen conditions.
The content of this RSS Feed (c) The Royal Society of Chemistry

10 Feb 09:21
Chem. Commun., 2021, 57,3070-3082
DOI: 10.1039/D1CC00528F, Feature Article
Open Access
Théo P. Gonçalves, Indranil Dutta, Kuo-Wei Huang
This feature article describes the recent conceptual understanding of aromaticity and its contribution to the thermodynamics in the catalytic process involving dearomatization and aromatization steps via metal-ligand cooperation.
The content of this RSS Feed (c) The Royal Society of Chemistry

10 Feb 07:37
Chem. Sci., 2021, 12,4484-4493
DOI: 10.1039/D0SC06406H, Edge Article
Open Access
Michelle P. van der Helm, Tuanke de Beun, Rienk Eelkema
We show, via simulations, how catalytic control over individual paths in a fuel-driven non-equilibrium chemical reaction network in batch or flow gives rise to responses in maximum conversion, lifetime and steady states.
The content of this RSS Feed (c) The Royal Society of Chemistry

10 Feb 07:36
by Robin F. Weitkamp,
Beate Neumann,
Hans‐Georg Stammler,
Berthold Hoge

Effect of hydrogen bonding on redox properties: The deprotonation of phenol derivatives with a tetraphosphazene base delivers non‐coordinated phenolates with zinc‐like redox potentials of up to −0.72(1) V vs. SCE, which are capable in radical anion synthesis and SF6 activation. The addition of phenol or water allows for the selective formation of phenolate adducts and enables the study of the effect of hydrogen bonding on phenolate redox properties.
Abstract
In this work, the syntheses of non‐coordinated electron‐rich phenolate anions via deprotonation of the corresponding alcohols with an extremely powerful perethyl tetraphosphazene base (Schwesinger base) are reported. The application of uncharged phosphazenes renders the selective preparation of anionic phenol‐phenolate and phenolate hydrates possible, which allows for the investigation of hydrogen bonding in these species. Hydrogen bonding brings about decreased redox potentials relative to the corresponding non‐coordinated phenolate anions. The latter show redox potentials of up to −0.72(1) V vs. SCE, which is comparable to that of zinc metal, thus qualifying their application as organic zinc mimics. We utilized phenolates as reducing agents for the generation of radical anions in addition to the corresponding phenoxyl radicals. A tetracyanoethylene radical anion salt was synthesized and fully characterized as a representative example. We also present the activation of sulfur hexafluoride (SF6) with phenolates in a SET reaction, in which the nature of the respective phenolate determines whether simple fluorides or pentafluorosulfanide ([SF5]−) salts are formed.
10 Feb 07:32
by Sunit Hazra, Koji Hirano, and Masahiro Miura

Organic Letters
DOI: 10.1021/acs.orglett.1c00050

10 Feb 07:30
by Li-Li Liao, Guang-Mei Cao, Yuan-Xu Jiang, Xing-Hao Jin, Xin-Long Hu, Jason J. Chruma, Guo-Quan Sun, Yong-Yuan Gui, and Da-Gang Yu

Journal of the American Chemical Society
DOI: 10.1021/jacs.0c11896

08 Feb 16:27
Chem. Commun., 2021, 57,2591-2604
DOI: 10.1039/D0CC08389E, Feature Article
Yan-Long Zheng, Stephen G. Newman
This feature article describes how diverse oxygen-containing functional groups such as esters, aldehydes, and alcohols can participate in cross-coupling reactions to prepare amides, ketones, alcohols, and beyond.
The content of this RSS Feed (c) The Royal Society of Chemistry

08 Feb 16:26
Green Chem., 2021, 23,1638-1641
DOI: 10.1039/D1GC00091H, Communication
Pannan Miao, Ruining Li, Xianfeng Lin, Liangming Rao, Zhankui Sun
Through a relay olefination and radical addition process, we developed cascade Wittig/hydroalkylation reactions induced by visible light. This metal-free radical approach features mild conditions, robustness, and excellent functionality tolerance.
The content of this RSS Feed (c) The Royal Society of Chemistry

08 Feb 16:26
by Hongxiang Li,
Honglei Liu,
Hongchao GUO

Abstract
Quaternary phosphonium salts have been extensively used in organic synthesis as Lewis acid organocatalysts. This review covers recent applications of phosphonium salts as Lewis acidic catalysts for Mannich, Strecker, and Friedel‐Crafts reactions for the formation of C−C bonds, annulation reactions, etc., allowing the construction of structurally diverse and synthetically useful architectures.
08 Feb 13:03
by Wei Shu,
Yi-Dan Du,
Bi-Hong Chen

A straightforward and modular route to a wide variety of aliphatic primary amines from alkenes is presented based on a metal‐free hydroamination at room temperature. The use of cost‐effective and easily available ammonium carbonate allows for the efficient conversion of terminal and internal alkenes with diverse substitution patterns into highly functionalized linear, α‐branched, and α‐tertiary primary amines.
Abstract
Direct and selective synthesis of primary amines from easily available precursors is attractive yet challenging. Herein, we report the rapid synthesis of primary amines from alkenes via metal‐free regioselective hydroamination at room temperature. Ammonium carbonate was used as ammonia surrogate for the first time, allowing for efficient conversion of terminal and internal alkenes into linear, α‐branched, and α‐tertiary primary amines under mild conditions. This method provides a straightforward and powerful approach to a wide spectrum of advanced, highly functionalized primary amines which are of particular interest in pharmaceutical chemistry and other areas.
08 Feb 11:31
by Carter N. Stout and Hans Renata

Accounts of Chemical Research
DOI: 10.1021/acs.accounts.0c00823

08 Feb 11:24
by Mahendra K. Sharma,
Dennis Rottschäfer,
Timo Glodde,
Beate Neumann,
Hans‐Georg Stammler,
Rajendra S. Ghadwal

A 1,4‐distannabenzene derivative 4 with two‐coordinated SnI atoms has been isolated as a green crystalline solid. The ground state of 4 is an open‐shell singlet (OS) with the singlet–triplet energy gap (ΔE
OS–T) of 4.4 kcal mol−1 (according to CASSCF, ΔE
S–T=6.6 kcal mol−1). Consequently, 4 exhibits a half‐field EPR signal at 100 K and undergoes H2 splitting at room temperature to quantitatively yield the SnII hydride 5 as an orange solid.
Abstract
The first SnI diradical [(ADCPh)Sn]2 (4) based on an anionic dicarbene (ADCPh={CN(Dipp)}2CPh; Dipp=2,6‐iPr2C6H3) scaffold has been isolated as a green crystalline solid by KC8 reduction of the corresponding bis‐chlorostannylene [(ADCPh)SnCl]2 (3). The six‐membered C4Sn2‐ring of 4 containing six π‐electrons shows a diatropic ring current, thus 4 may also be regarded as the first 1,4‐distannabenzene derivative. DFT calculations suggest an open‐shell singlet (OS) ground state of 4 with a remarkably small singlet–triplet energy gap (ΔEOS–T=4.4 kcal mol−1), which is consistent with CASSCF (ΔES–T=6.6 kcal mol−1 and diradical character y=37 %) calculations. The diradical 4 splits H2 at room temperature to yield the bis‐hydridostannylene [(ADCPh)SnH]2 (5). Further reactivity of 4 has been studied with PhSeSePh and MeOTf.










