08 Jul 15:22
by Baosheng Wei,
Qianyi Ren,
Thomas Bein,
Paul Knochel

A sequential cross‐coupling of benzal diacetates with organozinc reagents is achieved, delivering various functionalized triarylmethane and 1,1‐diarylalkane derivatives. This practical method features readily accessible substrates, transition‐metal‐free and one‐pot conditions, wide scope, scalability, and synthetic utility in the efficient synthesis of biologically relevant molecules.
Abstract
A variety of functionalized triarylmethane and 1,1‐diarylalkane derivatives were prepared via a transition‐metal‐free, one‐pot and two‐step procedure, involving the reaction of various benzal diacetates with organozinc reagents. A sequential cross‐coupling is enabled by changing the solvent from THF to toluene, and a two‐step SN1‐type mechanism was proposed and evidenced by experimental studies. The synthetic utility of the method is further demonstrated by the synthesis of several biologically relevant molecules, such as an anti‐tuberculosis agent, an anti‐breast cancer agent, a precursor of a sphingosine‐1‐phosphate (S1P) receptor modulator, and a FLAP inhibitor.
01 Mar 12:12
by Philip A. Provencher, Katherine L. Bay, John F. Hoskin, K. N. Houk, Jin-Quan Yu, and Erik J. Sorensen

ACS Catalysis
DOI: 10.1021/acscatal.0c05081

27 Feb 10:02
by Guangjun Li,
Xin Sui,
Xiao Cai,
Weigang Hu,
Xu Liu,
Mingyang Chen,
Yan Zhu

Tailor‐made catalytic sites for the chemical fixation of CO2 were achieved by implementing Ag atoms at different levels of liberation in atomically precise Au clusters. Au19Ag4(S‐Adm)15 with a completely open surface Ag site has the best catalytic activity, Au20Ag1(S‐Adm)15 with a partially open Ag site is least efficient, and Au21(S‐Adm)15 without a Ag site exhibits decent catalytic performance.
Abstract
Precise control of the composition and structure of active sites in an atom‐by‐atom fashion remains insuperable for heterogeneous catalysts. Here, we introduce tailor‐made catalytic sites for the cycloaddition of CO2 to epoxides achieved by implementing Ag atoms at different levels of liberation in atomically precise Au nanoclusters. Our results reveal that a single open Ag site on the Au19Ag4 cluster improves the ring‐opening of epoxides and sequent CO2 insertion, while the partially exposed Ag site on the Au20Ag1 cluster exhibits a weak affinity for epoxides and poor efficiency for CO2 capture. Structural tunability imparted by the atom‐by‐atom tailoring and unusual atomic charges distributed on Au and Ag atoms of the three clusters seem to be crucial for promoting challenging bond cleavages and formations in the chemical utilization of CO2.
27 Feb 09:59
by Lujuan Xu,
Seah Ling Kuan,
Tanja Weil

Site-selective dual protein functionalization serves as an invaluable tool for investigating protein structures and functions in complicated cellular environments and accomplishing semi-synthetic protein conjugates. The methodologies, as well as the applications of dual-functionalized protein bioconjugates are highlighted in this review.
Abstract
Site-selective protein functionalization serves as an invaluable tool for investigating protein structures and functions in complicated cellular environments and accomplishing semi-synthetic protein conjugates such as traceable therapeutics with improved features. Dual functionalization of proteins allows the incorporation of two different types of functionalities at distinct location(s), which greatly expands the features of native proteins. The attachment and crosstalk of a fluorescence donor and an acceptor dye provides fundamental insights into the folding and structural changes of proteins upon ligand binding in their native cellular environments. Moreover, the combination of drug molecules with different modes of action, imaging agents or stabilizing polymers provides new avenues to design precision protein therapeutics in a reproducible and well-characterizable fashion. This review aims to give a timely overview of the recent advancements and a future perspective of this relatively new research area. First, the chemical toolbox for dual functionalization of proteins is discussed and compared. The strengths and limitations of each strategy are summarized in order to enable readers to select the most appropriate method for their envisaged applications. Thereafter, representative applications of these dual-modified protein bioconjugates benefiting from the synergistic/additive properties of the two synthetic moieties are highlighted.
27 Feb 09:57
by Soumitra Agasti, Nicholas A. Beattie, Joseph J. W. McDouall, and David J. Procter

Journal of the American Chemical Society
DOI: 10.1021/jacs.1c01356

27 Feb 09:56
by Margarita Escudero-Casao, Giulia Licini, and Manuel Orlandi

Journal of the American Chemical Society
DOI: 10.1021/jacs.0c13236

26 Feb 12:38
by Ruben Martin,
Shang-Zheng Sun,
Laura Talavera,
Philipp Spiess,
Craig Day

A highly modular, site‐selective 1,1‐difunctionalization of unactivated olefins en route to versatile bis‐organometallic B,B(Si)‐reagents was developed by nickel catalyzed chain‐walking events. This protocol is characterized by exceptional reaction rates, mild conditions, broad scope, excellent chemo‐ and regioselectivity, thus unlocking a new technique for preparing densely functionalized 1,2‐bisorganometallic reagents from simple distinct, yet widely available, electrophilic partners.
Abstract
A catalytic 1,1‐difunctionalization of unactivated olefins en route to sp
3
bis‐organometallic B,B(Si)‐reagents is described. The protocol is characterized by exceptional reaction rates, mild conditions, wide scope, and exquisite selectivity pattern, constituting a new platform to access sp
3
bis‐organometallics.
26 Feb 10:58
by Natalie Holmberg-Douglas, Younggi Choi, Brian Aquila, Hoan Huynh, and David A. Nicewicz

ACS Catalysis
DOI: 10.1021/acscatal.1c00099

25 Feb 17:03
by Anastasia L. Gant Kanegusuku,
Jennifer L Roizen

This Review summarizes advances in photoredox-mediated Giese reactions since 2013, with a focus on the breadth of methods that provide access to crucial carbon-centered radical intermediates that can engage in radical conjugate addition processes.
Abstract
Photomediated Giese reactions are at the forefront of radical chemistry, much like the classical tin-mediated Giese reactions were nearly forty years ago. With the global recognition of organometallic photocatalysts for the mild and tunable generation of carbon-centered radicals, chemists have developed a torrent of strategies to form previously inaccessible radical intermediates that are capable of engaging in intermolecular conjugate addition reactions. This Review summarizes advances in photoredox-mediated Giese reactions since 2013, with a focus on the breadth of methods that provide access to crucial carbon-centered radical intermediates that can engage in radical conjugate addition processes.
25 Feb 15:10
Publication date: 26 March 2021
Source: Tetrahedron, Volume 84
Author(s): Togati Naveen
25 Feb 10:19
by Ming Chen,
Guangbin Dong

A diene‐platinum complex, (COD)Pt(TFA)2, serves as an efficient and chemoselective catalyst for α,β‐desaturation of cyclic ketones through direct Pt‐enolate formation. Distinct from known ketone dehydrogenation methods, a wide range of sensitive functional groups are tolerated. Mechanistic studies suggest a fast reversible α‐deprotonation followed by rate‐determining β‐H elimination.
Abstract
The development of a platinum‐catalyzed desaturation of cyclic ketones to their conjugated α,β‐unsaturated counterparts is reported in this full article. A unique diene‐platinum complex was identified to be an efficient catalyst, which enables direct metal‐enolate formation. The reaction operates under mild conditions without using strong bases or acids. Good to excellent yields can be achieved for diverse and complex scaffolds. A wide range of functional groups, including those sensitive to acids, bases/nucleophiles, or palladium species, are tolerated, which represents a distinct feature from other known desaturation methods. Mechanistically, this platinum catalysis exhibits a fast and reversible α‐deprotonation followed by a rate‐determining β‐hydrogen elimination process, which is different from the prior Pd‐catalyzed desaturation method. Promising preliminary enantioselective desaturation using a chiral‐diene‐platinum complex has also been obtained.
25 Feb 10:08
by Xiaojuan Li, Qiang Zhang, Weigang Zhang, Jinzhu Ma, Yi Wang and Yi Pan
Abstract
The difunctionalization of alkenes involving a trifluoromethylthio group (SCF3) for the conversion of versatile and readily available olefins into structurally more complex molecules has been successfully studied. However, the disproportionate dithiolation of alkenes is unknown. Herein, a transition-metal-free protocol is presented for the vicinal trifluoromethylthio–thiolation of unactivated alkenes via a radical process under mild conditions with a broad substrate scope and excellent tolerance.
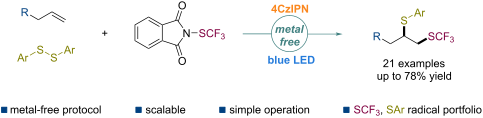
Beilstein J. Org. Chem. 2021, 17, 551–557. doi:10.3762/bjoc.17.49
24 Feb 13:00
Green Chem., 2021, 23,2553-2574
DOI: 10.1039/D0GC04040A, Critical Review
Robin Cauwenbergh, Shoubhik Das
This review presents an overview of the photoreduction of CO2 to formic acid using homogeneous catalysts.
The content of this RSS Feed (c) The Royal Society of Chemistry

24 Feb 11:33
by Sara E. Dibrell, Yujia Tao, and Sarah E. Reisman

Accounts of Chemical Research
DOI: 10.1021/acs.accounts.0c00858

24 Feb 11:18
by Arun Dixith Reddy Shada, Alexander J. M. Miller, Thomas J. Emge, and Alan S. Goldman

ACS Catalysis
DOI: 10.1021/acscatal.0c05160

24 Feb 10:57
by Geunho Choi,
Geun Seok Lee,
Beomsoon Park,
Dongwook Kim,
Soon Hyeok Hong

A photoinduced C(sp3)−H trifluoromethylation reaction of alkanes was developed by employing bpyCu(CF3)3 as a multifunctional reagent: photoinduced‐reaction initiator, precursor of the hydrogen‐atom‐transfer reagent, and trifluoromethyl anion source (see scheme). The operatively simple reaction enables the direct, late‐stage trifluoromethylation of complex molecules under mild reaction conditions.
Abstract
A mild and operationally simple C(sp3)−H trifluoromethylation method was developed for unactivated alkanes by utilizing a bench‐stable CuIII complex, bpyCu(CF3)3, as the initiator of the visible‐light photoinduced reaction, the source of a trifluoromethyl radical as a hydrogen atom transfer reagent, and the source of a trifluoromethyl anion for functionalization. The reaction was initiated by the generation of reactive electrophilic carbon‐centered CF3 radical through photoinduced homolytic cleavage of bpyCu(CF3)3, followed by hydrogen abstraction from an unactivated C(sp3)−H bond. Comprehensive mechanistic investigations based on a combination of experimental and computational methods suggested that C−CF3 bond formation was enabled by radical–polar crossover and ionic coupling between the resulting carbocation intermediate and the anionic CF3 source. The methylene‐selective reaction can be applied to the direct, late‐stage trifluoromethylation of natural products and bioactive molecules.
22 Feb 16:25
by Yuan Cai,
Lin‐Xin Ruan,
Abdul Rahman,
Shi‐Liang Shi

A method for general asymmetric addition of arylborons to simple ketones is reported, which is enabled by nickel/N‐heterocyclic carbene catalysis. Chiral tertiary alcohols are furnished with high efficiency and excellent levels of enantio‐ and chemocontrol, and the method offers a broad substrate scope.
Abstract
A general, efficient, highly enantio‐ and chemoselective N‐heterocyclic carbene (NHC)/Ni‐catalyzed addition of readily available and stable arylboronic esters to ketones is reported. This protocol provides unexpectedly fast access (usually 10 min) to various chiral tertiary alcohols with exceptionally broad substrate scope and excellent functional group tolerance (76 examples, up to 98 % ee). This process is orthogonal to other known Ni‐mediated Suzuki–Miyaura couplings, as it tolerates aryl chlorides, fluorides, ethers, esters, amides, nitriles, and alkyl chlorides. The reaction is applied to late‐stage modifications of various densely functionalized medicinally relevant molecules. Preliminary mechanistic studies suggest that a rare enantioselective η2‐coordinating activation of ketone carbonyls is involved. This cross‐coupling‐like mechanism is expected to enable other challenging transformations of ketones.
19 Feb 16:11
Chem. Sci., 2021, 12,2016-2024
DOI: 10.1039/D0SC03278F, Minireview

Open Access
Abir Sarbajna, V. S. V. S. N. Swamy, Viktoria H. Gessner
The application of ylide substituents as strong donor ligands for the stabilization of reactive main group compounds with unusual properties and reactivities is discussed.
The content of this RSS Feed (c) The Royal Society of Chemistry

19 Feb 16:10
Org. Biomol. Chem., 2021, 19,2312-2321
DOI: 10.1039/D1OB00014D, Paper
Open Access
Dušan Kolarski, Akiko Sugiyama, Theo Rodat, Albert Schulte, Christian Peifer, Kenichiro Itami, Tsuyoshi Hirota, Ben L. Feringa, Wiktor Szymanski
6-Azopurines were evaluated for their reductive stability, and the ability to modulate CKIα activity and cellular circadian rhythms, revealing key challenges for long-term activity modulation utilizing chronophotopharmacology.
The content of this RSS Feed (c) The Royal Society of Chemistry

19 Feb 16:06
by Huaquan Fang,
Kaixue Xie,
Sebastian Kemper,
Martin Oestreich

Acyclic tertiary amines with alkyl substitution undergo two consecutive C(sp3)−H silylation reactions with dihydrosilanes to form 4‐silapiperidines. Bond formation occurs β to the nitrogen atom at two of the alkyl residues. The reaction is catalyzed by the strong boron Lewis acid B(C6F5)3 and involves enamine intermediates generated by dehydrogenation.
Abstract
Tris(pentafluorophenyl)borane has been found to catalyze the two‐fold C(sp3)−H silylation of various trialkylamine derivatives with dihydrosilanes, furnishing the corresponding 4‐silapiperidines in decent yields. The multi‐step reaction cascade involves amine‐to‐enamine dehydrogenation at two alkyl residues and two electrophilic silylation reactions of those enamines, one inter‐ and one intramolecular.
19 Feb 16:06
by Vanda Dašková,
Jeffrey Buter,
Anne K Schoonen,
Martin Lutz,
Folkert de Vries,
Ben L Feringa

The origin of homochirality is a fundamental scientific question. We show that dissolution behavior of racemic and enantiopure amines can be altered by conversion into phosphoramidates. Stirring a solid scalemic mixture in water leads to enantioenrichment of the solution phase (>90 % ee). Amplification of amino acids and their derivatives show that phosphorylation can serve as a potentially viable source of prebiotic homochirality.
Abstract
The origin of biomolecular homochirality continues to be one of the most fascinating aspects of prebiotic chemistry. Various amplification strategies for chiral compounds to enhance a small chiral preference have been reported, but none of these involves phosphorylation, one of nature's essential chemical reactions. Here we present a simple and robust concept of phosphorylation‐based chiral amplification of amines and amino acids in water. By exploiting the difference in solubility of a racemic phosphoramidate and its enantiopure form, we achieved enantioenrichment in solution. Starting with near racemic, phenylethylamine‐based phosphoramidates, ee's of up to 95 % are reached in a single amplification step. Particularly noteworthy is the enantioenrichment of phosphorylated amino acids and their derivatives, which might point to a potential role of phosphorus en‐route to prebiotic homochirality.
19 Feb 16:05
by Suman Chakrabarty, Evan O. Romero, Joshua B. Pyser, Jessica A. Yazarians, and Alison R. H. Narayan

Accounts of Chemical Research
DOI: 10.1021/acs.accounts.0c00810

18 Feb 11:37
by Samir Isaac Meramo Hurtado, Plinio Puello, and Amaury Cabarcas

ACS Omega
DOI: 10.1021/acsomega.0c06088
17 Feb 08:03
by Lan Zheng, Yu-En Qian, Yuan-Zhuo Hu, Jun-An Xiao, Zhi-Peng Ye, Kai Chen, Hao-Yue Xiang, Xiao-Qing Chen, and Hua Yang
![TOC Graphic]()
Organic Letters
DOI: 10.1021/acs.orglett.1c00064

17 Feb 08:02
by Koushik Sarkar, Kuhali Das, Abhishek Kundu, Debashis Adhikari, and Biplab Maji

ACS Catalysis
DOI: 10.1021/acscatal.0c05406

17 Feb 08:02
by Michael Rauch, Jie Luo, Liat Avram, Yehoshoa Ben-David, and David Milstein

ACS Catalysis
DOI: 10.1021/acscatal.1c00418

17 Feb 08:01
by Guan-Wen Yang, Cheng-Kai Xu, Rui Xie, Yao-Yao Zhang, Xiao-Feng Zhu, and Guang-Peng Wu

Journal of the American Chemical Society
DOI: 10.1021/jacs.0c12425

16 Feb 15:42
by Nate W. J. Ang,
Lutz Ackermann

Nickela‐electrothiolation: A nascent co‐operation between nickel catalysis and electrochemical synthesis to bring forth cross‐electrophile thiolation of alkyl bromides.
Abstract
Sulfur‐containing molecules are of utmost topical importance towards the effective development of pharmaceuticals and functional materials. Herein, we present an efficient and mild electrochemical thiolation by cross‐electrophile coupling of alkyl bromides with functionalized bench‐stable thiosulfonates to access alkyl sulfides with excellent efficacy and broad functional group tolerance. Cyclic voltammetry and potentiostatic analysis were performed to elucidate mechanistic insights into this electrocatalytic thiolation reaction.
16 Feb 09:37
by Timothy D. Schoch, Mukulesh Mondal, and Jimmie D. Weaver
![TOC Graphic]()
Organic Letters
DOI: 10.1021/acs.orglett.0c04305

16 Feb 09:34
by Bing Tian
Nature Catalysis, Published online: 15 February 2021; doi:10.1038/s41929-021-00574-5
Asymmetric dioxygenation of alkenes is an attractive concept to synthesize enantioenriched diols, but the performance in the case of terminal alkenes is low with current methods. Now, this has been addressed by an asymmetric oxypalladation process that provides access to enantioenriched 1,2-diols, enabled by the chiral 6-modified pyridinyl oxazoline ligand.





