
ACS Catalysis
DOI: 10.1021/acscatal.2c02410



Nature Chemistry, Published online: 01 July 2022; doi:10.1038/s41557-022-00993-2
Antibody-mediated delivery of therapeutics has been primarily limited to agents containing amine, alcohol or thiol functional groups. Now, an approach has been developed to create stable and bio-reversible prodrugs that mask ortho-quinones. Drug release requires both protease activation followed by acid-assisted elimination.
A visible-light-induced nickel/photoredox-catalyzed mono-Suzuki–Miyaura cross-coupling of 1,2-bis-boronic esters has been accomplished, which is selective for the most sterically hindered boronic ester. High regioselectivities were achieved through a facile radical 1,2-boron shift process, wherein the initially generated primary β-boryl radical is rapidly converted to the thermodynamically more stable secondary radical before nickel-catalyzed cross-coupling.
Site-selective transition-metal-catalyzed mono-deboronative cross-couplings of 1,2-bis-boronic esters are valuable methods for the synthesis of functionalized organoboron compounds. However, such cross-couplings are limited to reaction of the sterically less hindered primary boronic ester. Herein, we report a nickel/photoredox-catalyzed mono-deboronative arylation of 1,2-bis-boronic esters that is selective for coupling of the more sterically hindered secondary/tertiary position. This is achieved by taking advantage of a 1,2-boron shift of primary β-boryl radicals to the thermodynamically favored secondary/tertiary radicals, which are subsequently intercepted by the nickel catalyst to enable arylation. The mild conditions are amenable to a broad range of aryl halides to give β-aryl boronic ester products in good yields and with high regioselectivity. This method also allows stereodivergent coupling of cyclic cis-1,2-bis-boronic esters to give trans-substituted products.

A series of supported MoO3 catalysts were prepared and investigated for the selective demethoxylation of guaiacol. A 5% MoO3/TiO2 (P25) catalyst showed the best performance for guaiacol demethoxylation, and was also successfully applied for the demethoxylation of 4-n-propylguaiacol and a bioliquid enriched in guaiacols. Characterization of the MoO3/TiO2 (P25) catalysts reveals that a high surface area, a high amount of weak and medium acid sites, and an intermediate oxygen vacancy concentration are preferred for a high selectivity to demethoxylated products.
Lignin-derived monomers with methoxy substituents are abundantly present in bioliquids derived from lignocellulosic biomass. Examples are the products obtained from the reductive catalytic fractionation of lignin (RCF) and pyrolysis of lignocellulosic biomass and hydrotreated products thereof. An attractive valorization step for these liquids involves demethoxylation to obtain alkylated phenols through selective catalytic hydrodeoxygenation (HDO). Within the context of sustainable chemistry, there is a strong drive to use cheap, non-precious metal catalysts for this purpose. In this study, the HDO of guaiacol (5 wt% in toluene) was investigated in a continuous fixed-bed reactor at 380 °C, 20 bar over supported MoO3 catalysts. MoO3 (5 %) supported on TiO2 (P25) was shown to give superior performance compared with MoO3 supported on anatase TiO2, Al2O3, SiO2, Nb2O5, CeO2, and ZrO2. Additional studies involving variation of the Mo loading and process conditions were performed, and the highest selectivity to demethoxylated phenolics like phenol and methylated phenols was 82 % at 97 % conversion of guaiacol. Both 4-n-propylguaiacol and a realistic guaiacols-rich feed isolated from a representative pyrolysis oil were also successfully demethoxylated with the 5 % MoO3/TiO2 catalyst.

Nature, Published online: 28 June 2022; doi:10.1038/d41586-022-01806-9
People with similar body odours are more likely to instantly get along as friends. Plus, China gets closer to its first mRNA vaccine and how to make spatial maps of gene activity — down to the cellular level.
Full-spectrum photocatalysis is believed to play an important role in efficient utilization of solar light. Recent progresses on full-solar-light-driven photocatalytic systems are outlined. The existing challenges and prospects of full-spectrum photocatalysis are also discussed.
The ability to use the full solar spectrum energy in photocatalytic processes for environmental remediation and energy production has attracted worldwide attention. Efficient harvesting of near-infrared (NIR) photons, especially in the wavelength range beyond 800 nm, is one of the main driving forces in photocatalytic research. The design of appropriate photocatalytic systems for wide-range light-harvesting from the UV to NIR regions is a promising method of maximizing the efficiency of solar energy utilization. This review comprehensively summarizes the recent progress in full-solar-light-driven photocatalytic systems, including several strategies to harness NIR light and thermal energy. The corresponding photocatalytic mechanisms and design of binary or ternary heterogeneous systems are discussed in detail. Moreover, future perspectives and challenges are presented to inspire the development of further innovations in full-solar-light-driven photocatalysis.

Hydrogenation catalysis: A homogeneous Cu(I) catalyst with high selectivity (>99%) and activity (TON=4 000), under relatively mild reaction conditions (24 h, 140 °C) using a base-free system using H2 (300 psi) as a reductant, and a low catalytic load (0.025 mol%) under optimized conditions in the hydrogenation of levulinic acid to γ-valerolactone is reported, along with full characterization of the copper catalytic precursor.
We report the first well-defined homogeneous copper-based catalyst for levulinic acid (LVA) hydrogenation using complex [(PPh3)2Cu(k2-O,O-LVA)] as a copper source and (1,2-bis(diisopropyl phosphino)ethane (dippe) as an ancillary ligand. This catalytic precursor has high activity in LVA hydrogenation and yields γ-valerolactone (GVL) under relatively mild reaction conditions (24 h, 140 °C) in a base-free system using H2 (300 psi) as a reductant. Under optimized conditions, GVL was obtained with excellent yield (>99 %) and selectivity (>99 %) using a low catalytic load (0.025 mol %), allowing a TON=4000.
Nature Chemistry, Published online: 27 June 2022; doi:10.1038/s41557-022-00973-6
Despite mechanically axially chiral (MAC) catenanes being recognized in 1961, their stereoselective synthesis had not been disclosed until now. Closer inspection of the MAC stereogenic unit has also led to the identification of an analogous, but unremarked upon, form of rotaxane stereochemistry and the conceptualization of a general approach to prepare MAC molecules stereoselectively. Open Access
Open Access   This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.

This review outlines a handful of transformations employing transition-metal and photoredox catalysis by auxiliary-assisted C−H activation strategy to assemble synthetic libraries of C−C and C-heteroatom bonds. The review also features the substrate scope and mechanistic insights of the key methodologies.
Metallaphotoredox catalysis represents the combination of two activation modes: metal-catalyzed C−H functionalization and visible-light-induced photocatalysis. This appealing dual catalysis technique has evolved as a versatile platform that has paved the way for diverse low-energy pathways for a plethora of synthetic transformations. The synergistic combination of two “green” approaches has garnered enormous interest owing to the development of sustainable strategies. In the recent past, significant advancements have been accomplished in constructing site-selective C−C and C-heteroatom bonds. This review outlines the use of photoredox catalysis in directing group-assisted C−H functionalization reactions. The mechanistic insights of the developments and synthetic applications are addressed.
![The Many Chemists Who Could Have Proposed the Woodward-Hoffmann Rules But Didn't: The Organic Chemists Who Discovered the Smoking Guns[]**](https://onlinelibrary.wiley.com/cms/asset/bb74475d-fa8e-4721-ac0b-1754e3ee9983/tcr202200065-toc-0001-m.png)
Emanuel Vogel, Rudolf Criegee, and William G. Dauben were three of the organic chemists in whose laboratories some of the earliest experimental hints were observed for the mechanism of the no-mechanism reaction.
It is a reasonable question to ask, why, as of 1965 when the five Woodward-Hoffmann communications appeared, did no other organic chemist discover the orbital symmetry rules for pericyclic reactions? Two theoretical chemists – Luitzen Oosterhoff (in 1961) and Kenichi Fukui (in 1964) had discovered portions of the orbital symmetry rules before Woodward and Hoffmann. Why not organic chemists? Indeed, perhaps the greatest motivation to discover the mechanism of a mysterious reaction is to uncover key examples of that mysterious reaction in your very own laboratory. The stories of 20 chemists and R. B. Woodward are discussed in this paper which is Paper 6 in a 27-paper series on the history of Woodward-Hoffmann rules. Social, political, and scientific explanations will also be presented as partial explanations as to why none of these individuals – except Woodward with Hoffmann – solved the pericyclic no-mechanism problem.
Nature, Published online: 22 June 2022; doi:10.1038/d41586-022-01701-3
These extinct uber-predators occupied an ecological niche that has no known equivalent in the modern ocean.Synlett
DOI: 10.1055/a-1863-9090

In the absence of transition-metal catalysts and additional oxidants, arylhydroxylamines can undergo an unconventional O-sulfinylation/[2,3]-sigmatropic rearrangement/rearomatization cascade with trifluoromethylsulfinyl chloride to rapidly and efficiently synthesize versatile ortho-sulfonylated aromatic amines. This Synpacts article describes the discovery, further study, and practical application of the rearrangement reactions in arylhydroxylamine compounds and highlights our recent advances in this area.1 Introduction2 Discovery of a New Reaction3 O-Sulfinylation of Arylhydroxylamines Followed by [2,3]-σ Rearrangement4 Conclusion
[...]
Georg Thieme Verlag KG Rüdigerstraße 14, 70469 Stuttgart, Germany
Article in Thieme eJournals:
Table of contents | Abstract | Full text
Abstract
Isocyanides are hardly produced, dramatically sensitive to purification processes, and complex to handle as synthetic tools. Notwithstanding, they represent one of the most refined and valuable compounds for accessing sophisticated and elegant synthetic routes. A unique interest has always been addressed to their production, though their synthetic pathways usually involve employing strong conditions and toxic reagents. The current paper intends to provide a conceptually innovative synthetic protocol for mechanochemical isocyanide preparation, simultaneously lowering the related reagents' toxicity and improving their purification in a straightforward procedure.
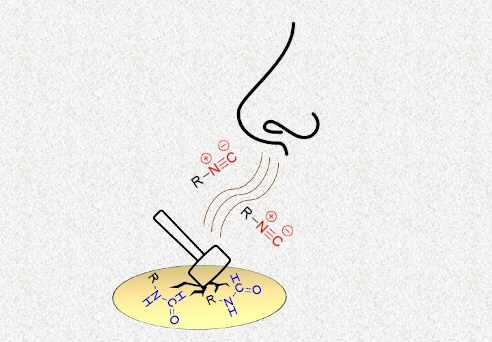
Beilstein J. Org. Chem. 2022, 18, 732–737. doi:10.3762/bjoc.18.73


Open Access  This article is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence.
Publication date: August 2022
Source: Trends in Chemistry, Volume 4, Issue 8
Author(s): Daily Rodriguez-Padron, Awais Ahmad, Pablo Romero-Carrillo, Rafael Luque, Roberto Esposito


A transition-metal-free aminocyanation of aryl alkynes has been achieved using indium tribromide, InBr3 or B(C6F5)3 as a Lewis acid. This aminocyanation protocol features with non-toxic cyanide source, a good substrate scope and potentially valuable aminocyanation products. Mechanistic studies reveal the complex formation between Lewis acid and alkyne to produce in situ alkyne nitrile as a key intermediate. Further hydroamination of alkyne nitrile with arylamines affords the E-selective (E:Z=70:30 to 90:10) β-aminoacrylonitrile derivatives.

Two for one: Dual-atom catalysts (DACs) have shown excellent performances in catalysis. This Review summarizes the research progresses of homonuclear and heteronuclear DACs and their applications in oxygen reduction, carbon dioxide reduction, hydrogen evolution, oxygen evolution, Zn-air batteries, tandem catalytic reactions, and so on. Moreover, the challenges and perspectives of DACs are put forward.
Dual-atom catalysts (DACs) are an important branch of single-atom catalysts (SACs), in which the former can effectively break the dilemma faced by the traditional SACs. The synergetic effects between bimetallic atoms provide many active sites, promising to improve catalytic performance and even catalyze more complex reactions. This paper reviews the recent research progresses of two kinds of DACs, including homonuclear and heteronuclear DACs, and their applications in oxygen reduction, carbon dioxide reduction, hydrogen evolution, oxygen evolution, Zn-air batteries, tandem catalytic reactions, and so on. In addition, in order to promote the further development of DACs, the challenges and perspectives of DACs are put forward.

Power play: The Power-to-X synthesis of acetic acid by formal isomerization of the renewable carbon source methyl formate (MF) is realized applying a homogeneous molecular system. The base-catalyzed decarbonylation of MF, yielding CO and methanol in situ, is combined with the synthesis of acetic acid and methyl acetate with Pd catalysts (turnover number up to 43000, acetate selectivity up to 83 %).
The synthesis of acetic acid by formal isomerization of methyl formate (MF) was investigated using molecular catalysts. The base-catalyzed decarbonylation of MF, yielding CO and methanol in situ, was integrated with their palladium-catalyzed recombination for the synthesis of acetic acid and methyl acetate in a one pot reaction. The complex [Pd(Cl)2(dppe)] [dppe=1,2-bis(diphenylphosphino)-ethane] in combination with NaI as iodide source and NaOMe as base were identified as promising molecular components to enable the overall conversion. Sequential application of the statistical methods design of experiments and simplex optimization was used in combination with thermodynamic analysis of the competing reaction pathways for experimental planning and data analysis. Starting from a proof-of-principle with a turnover number (TON) of 11, the catalytic system could thus be optimized to allow quantitative conversion of MF with a TON of 43000, whereby a yield of 83 % of acetate groups and a yield of 74 % for free acetic acid was obtained.





