06 Mar 12:14
by Zi-Yu Li, Zhen Yuan, Yan-Xia Zhao, Sheng-Gui He
Abstract

06 Mar 12:09
by Matthew N. Hopkinson, Basudev Sahoo, Jun-Long Li, Frank Glorius
Abstract
The photoredox activation of organic substrates with visible light is a powerful methodology that generates reactive radical species under very mild conditions. When combined with another catalytic process in a dual catalytic system, novel, visible-light-promoted transformations have been realized that do not proceed using either catalyst in isolation. In this minireview, the state of the art in organic reactions mediated by dual catalytic systems merging photoredox activation with organo-, acid or metal catalysis is discussed.

De(light)ful catalysis! The merger of photoredox catalysis with another catalytic mode can result in novel, visible-light-promoted reactions that do not proceed by using either catalyst independently. Herein, the different ways that two catalytic modes can operate in tandem are highlighted, focusing on dual-catalyzed organic processes that merge photoredox with organo-, acid, and transition-metal catalysis.
06 Mar 11:55
by Yamil J. Colón, David Fairen-Jimenez, Christopher E. Wilmer and Randall Q. Snurr

The Journal of Physical Chemistry C
DOI: 10.1021/jp4122326

06 Mar 11:53
by Richard J. Burford, Warren E. Piers, Daniel H. Ess and Masood Parvez

Journal of the American Chemical Society
DOI: 10.1021/ja412650s

06 Mar 10:16
by Sergey A. Denisov, Yanouk Cudré, Peter Verwilst, Gediminas Jonusauskas, Marta Marín-Suárez, Jorge Fernando Fernández-Sánchez, Etienne Baranoff and Nathan D. McClenaghan

Inorganic Chemistry
DOI: 10.1021/ic4030712

06 Mar 10:15
by Daniel L. DuBois

Inorganic Chemistry
DOI: 10.1021/ic4026969

06 Mar 10:12
by Jonas Sundberg, Hannes Witt, Lisa Cameron, Mikael Håkansson, Jesper Bendix and Christine J. McKenzie

Inorganic Chemistry
DOI: 10.1021/ic402599e

06 Mar 10:11
by Yohei Hattori, Michihiro Nishikawa, Tetsuro Kusamoto, Shoko Kume and Hiroshi Nishihara

Inorganic Chemistry
DOI: 10.1021/ic500074c

06 Mar 10:06
by Christa Lübbe, Andreas Dumrath, Helfried Neumann, Marion Schäffer, Ralf Zimmermann, Matthias Beller, Renat Kadyrov
Abstract
The dramatic effect of substrate impurities on the performance of a specific ruthenium catalyst system is demonstrated in the benchmark metathesis reaction of diethyl diallylmalonate. Based on detailed two-dimensional GC–time-of-flight MS measurements, the significant influence of small amounts of different contaminations, especially various organic halides, is shown. This work serves as an incisive example of the importance of impurities to catalyst performance, also in homogeneous catalysis, which is often ignored in academic research.

Pure or not pure, that is the question: The presence of small amounts of impurities has a dramatic influence on the reactivity and selectivity of a novel ruthenium-based ring-closing metathesis (RCM) catalyst.
06 Mar 10:05
by Ning Wang, Zhenxin Xu, Jie Deng, Kui Shen, Xiaopeng Yu, Weizhong Qian, Wei Chu, Fei Wei
Abstract
Ordered mesoporous NiAl and NiCeAl catalysts with different Ce/Al molar ratios were facilely synthesized by using the improved evaporation-induced self-assembly method. The characterization results confirmed that the ordered mesoporous structure was well sustained in the Ce-incorporated NiAl materials (Ce/Al molar ratio<4 %). Compared with NiAl mesoporous materials, Ce-incorporated mesoporous materials demonstrated higher specific surface areas, larger pore volumes, and more uniform pore sizes. The catalytic test conducted by using methane dry reforming revealed that compared with NiAl catalysts, all the Ce-promoted catalysts demonstrated improved initial catalytic activity, which was due to the high dispersion and the high reduction degree of active Ni species in NiCeAl catalysts. The stable alumina framework, confinement effect of ordered mesopores, and high oxygen mobility contributed to the improved catalytic stability of NiCeAl catalysts. For comparison, the mesoporous NiCeAl catalyst (denoted as NiCeAl-IMP) was also prepared by using the conventional impregnation method. The agglomeration of Ni particles was observed during the stability test for the Ni-impregnated catalyst, which accelerated the rate of carbon deposition. The NiCeAl catalyst (Ce/Al molar ratio=1 %) demonstrated excellent resistance to the formation of graphitic carbon species owing to the redox property, while a large amount of graphitic carbon species was deposited over the NiAl sample, which was responsible for deactivation.

Everything begins with an order: Ordered mesoporous NiAl and NiCeAl (R) catalysts with various Ce contents were prepared by using the improved evaporation-induced self-assembly method. Compared with NiAl catalysts, Ce-incorporated catalysts demonstrated higher specific surface areas, larger pore volumes, and more uniform pore sizes. The incorporation of Ce promoted the high dispersion and high reducibility of Ni species, which led to an improved catalytic activity for methane dry reforming.
06 Mar 10:03
by Jakob Kibsgaard

Nature Chemistry 6, 248 (2014).
doi:10.1038/nchem.1853
Authors: Jakob Kibsgaard, Thomas F. Jaramillo & Flemming Besenbacher
Non-noble-metal-based MoS2 nanostructures are hydrogen evolution catalysts whose active sites are known to be located at the edges. Supported thiomolybdate [Mo3S13]2− nanoclusters have now been prepared that exhibit a structural motif similar to that of MoS2 edges. The nanoclusters, synthesized by a scalable route, demonstrate a high turnover frequency.
06 Mar 10:01
by Ruth Doherty
Nature Chemistry 6, 168 (2014).
doi:10.1038/nchem.1884
Author: Ruth Doherty
06 Mar 09:50
Chem. Soc. Rev., 2014, 43,3480-3524
DOI: 10.1039/C3CS60282F, Review Article
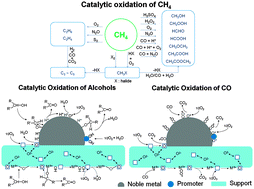
Zhen Guo, Bin Liu, Qinghong Zhang, Weiping Deng, Ye Wang, Yanhui Yang
Selective oxidations of CH4, alcohols and CO over heterogeneous catalysts are reviewed from the viewpoint of green and sustainable chemistry.
The content of this RSS Feed (c) The Royal Society of Chemistry

gjchen and -1 others like this
06 Mar 09:49
Chem. Soc. Rev., 2014, 43,7838-7869
DOI: 10.1039/C3CS60409H, Review Article
Suojiang Zhang, Jian Sun, Xiaochun Zhang, Jiayu Xin, Qingqing Miao, Jianji Wang
We review ionic liquid-based processes in the renewable energy field, including CO2 conversion, biomass conversion, solar energy and energy storage.
The content of this RSS Feed (c) The Royal Society of Chemistry

06 Mar 09:48
Chem. Soc. Rev., 2014, 43,7562-7580
DOI: 10.1039/C3CS60396B, Tutorial Review
Paola Lanzafame, Gabriele Centi, Siglinda Perathoner
The use of biomass, bio-waste and CO2 derived raw materials, the latter synthesized using H2 produced using renewable energy sources, opens new scenarios to develop a sustainable and low carbon chemical production.
The content of this RSS Feed (c) The Royal Society of Chemistry

06 Mar 09:48
by Wolfgang Grünert, Dennis Großmann, Heshmat Noei, Marga-Martina Pohl, Ilya Sinev, Andrea De Toni, Yuemin Wang, Martin Muhler
Abstract
Au/TiO2 catalysts prepared by a deposition–precipitation process and used for CO oxidation without previous calcination exhibited high, largely temperature-independent conversions at low temperatures, with apparent activation energies of about zero. Thermal treatments, such as He at 623 K, changed the conversion–temperature characteristics to the well-known S-shape, with activation energies slightly below 30 kJ mol−1. Sample characterization by XAFS and electron microscopy and a low-temperature IR study of CO adsorption and oxidation showed that CO can be oxidized by gas-phase O2 at 90 K already over the freeze-dried catalyst in the initial state that contained Au exclusively in the +3 oxidation state. CO conversion after activation in the feed at 303 K is due to AuIII-containing sites at low temperatures, while Au0 dominates conversion at higher temperatures. After thermal treatments, CO conversion in the whole investigated temperature range results from sites containing exclusively Au0.

Ionic or metallic: Au3+ ions on TiO2 (see HAADF-STEM image of a freshly prepared sample) can catalyze the oxidation of CO at low temperatures. The reaction rates at Au3+-containing centers are similar to those found at metallic gold clusters. However, the apparent activation energies are very low, which is probably due to the opposing influence of the true activation energy and the adsorption enthalpy of CO on Au3+ centers.
06 Mar 09:09
by Axel G. Griesbeck

John Wiley and Sons, Hoboken, 2013. 380 pp., hardcover, € 132.00.—ISBN 978-0470915349
06 Mar 09:08
by Jean-Cyrille Hierso

Chemical Reviews
DOI: 10.1021/cr400330g

06 Mar 09:04
by Edward I. Solomon, David E. Heppner, Esther M. Johnston, Jake W. Ginsbach, Jordi Cirera, Munzarin Qayyum, Matthew T. Kieber-Emmons, Christian H. Kjaergaard, Ryan G. Hadt and Li Tian

Chemical Reviews
DOI: 10.1021/cr400327t

18 Feb 11:41
by Tatsuhiko Mukuta, Naoto Fukazawa, Kei Murata, Akiko Inagaki, Munetaka Akita, Sei’ichi Tanaka, Shin-ya Koshihara and Ken Onda

Inorganic Chemistry
DOI: 10.1021/ic402474t

18 Feb 11:38
by Jun Xing, Jian Fu Chen, Yu Hang Li, Wen Tao Yuan, Ying Zhou, Li Rong Zheng, Hai Feng Wang, P. Hu, Yun Wang, Hui Jun Zhao, Yong Wang, Hua Gui Yang

TiO2 photocatalysts loaded with isolated Pt atoms were successfully synthesized by a simple and convenient technique as described by H. F. Wang and H. G. Yang et al. in their Communication on page 2138 ff. Isolated Pt atoms can stably anchor on TiO2 and exhibit a high photocatalytic hydrogen evolution performance compared with Pt nanoparticles or clusters. Moreover, the configurations of the isolated Pt atoms and their catalytic hydrogen evolution activity were calculated by large-scale periodic DFT analysis. The results obtained for such model catalysts not only opens a door for synthesizing high-efficiency catalytic materials, but also has a great potential to reduce the high cost of commercial noble metal catalysts in industry.
18 Feb 11:37
by Cristiano Zuccaccia, Gianfranco Bellachioma, Olga Bortolini, Alberto Bucci, Arianna Savini, Alceo Macchioni
Abstract
The reaction of [Cp*Ir(bzpy)NO3] (1; bzpy=2-benzoylpyridine, Cp*=pentamethylcyclopentadienyl anion), a competent water-oxidation catalyst, with several oxidants (H2O2, NaIO4, cerium ammonium nitrate (CAN)) was studied to intercept and characterize possible intermediates of the oxidative transformation. NMR spectroscopy and ESI-MS techniques provided evidence for the formation of many species that all had the intact Ir–bzpy moiety and a gradually more oxidized Cp* ligand. Initially, an oxygen atom is trapped in between two carbon atoms of Cp* and iridium, which gives an oxygen–Ir coordinated epoxide, whereas the remaining three carbon atoms of Cp* are involved in a η3 interaction with iridium (2 a). Formal addition of H2O to 2 a or H2O2 to 1 leads to 2 b, in which a double MeCOH functionalization of Cp* is present with one MeCOH engaged in an interaction with iridium. The structure of 2 b was unambiguously determined in the solid state and in solution by X-ray single-crystal diffractometry and advanced NMR spectroscopic techniques, respectively. Further oxidation led to the opening of Cp* and transformation of the diol into a diketone with one carbonyl coordinated at the metal (2 c). A η3 interaction between the three non-oxygenated carbons of “ex-Cp*” and iridium is also present in both 2 b and 2 c. Isolated 2 b and mixtures of 2 a–c species were tested in water-oxidation catalysis by using CAN as sacrificial oxidant. They showed substantially the same activity than 1 (turnover frequency values ranged from 9 to 14 min−1).

Signs of three: Three intermediates from the oxidative transformation of a Cp*–iridium water-oxidation catalyst have been intercepted and characterized by using NMR spectroscopy, ESI-MS and, in one case, X-ray crystallography. Progressive oxidation of Cp* has been observed, whereas the benzoylpyridine ancillary ligand remains intact. Isolated intermediates and their mixture are still active in water-oxidation catalysis (see scheme).
18 Feb 11:35
by Frédéric-Georges Fontaine, Marc-André Courtemanche, Marc-André Légaré
Abstract
Metal-free systems, including frustrated Lewis pairs (FLPs) have been shown to bind CO2. By reducing the Lewis acidity and basicity of the ambiphilic system, it is possible to generate active catalysts for the deoxygenative hydroboration of carbon dioxide to methanol derivatives with conversion rates comparable to those of transition-metal-based catalysts.

Less is more: Metal-free systems, including frustrated Lewis pairs (FLPs), have been shown to bind CO2. By reducing the Lewis acidity and basicity of the ambiphilic system, it is possible to generate active catalysts for the deoxygenative hydroboration of carbon dioxide to methanol derivatives with conversion rates comparable to those of transition-metal-based catalysts (see scheme).
18 Feb 11:19
by Jordan J. Stracke and Richard G. Finke

ACS Catalysis
DOI: 10.1021/cs4011716

12 Feb 08:22
by Rodney D. L. Smith, Barbora Sporinova, Randal D. Fagan, Simon Trudel and Curtis P. Berlinguette

Chemistry of Materials
DOI: 10.1021/cm4041715

12 Feb 08:22
by Dirk Hollmann, Michael Karnahl, Stefanie Tschierlei, Kamalakannan Kailasam, Matthias Schneider, Jörg Radnik, Kathleen Grabow, Ursula Bentrup, Henrik Junge, Matthias Beller, Stefan Lochbrunner, Arne Thomas and Angelika Brückner

Chemistry of Materials
DOI: 10.1021/cm500034p

12 Feb 08:19
by Meysam Pazoki, Nima Taghavinia, Anders Hagfeldt and Gerrit Boschloo

The Journal of Physical Chemistry C
DOI: 10.1021/jp4113574

12 Feb 08:13
by Rik V. Mom, Jun Cheng, Marc T. M. Koper and Michiel Sprik

The Journal of Physical Chemistry C
DOI: 10.1021/jp409373c

10 Feb 10:33
by Sarina Sarina, Huai-Yong Zhu, Qi Xiao, Esa Jaatinen, Jianfeng Jia, Yiming Huang, Zhanfeng Zheng, Haishun Wu
Abstract
Supported nanoparticles (NPs) of nonplasmonic transition metals (Pd, Pt, Rh, and Ir) are widely used as thermally activated catalysts for the synthesis of important organic compounds, but little is known about their photocatalytic capabilities. We discovered that irradiation with light can significantly enhance the intrinsic catalytic performance of these metal NPs at ambient temperatures for several types of reactions. These metal NPs strongly absorb the light mainly through interband electronic transitions. The excited electrons interact with the reactant molecules on the particles to accelerate these reactions. The rate of the catalyzed reaction depends on the concentration and energy of the excited electrons, which can be increased by increasing the light intensity or by reducing the irradiation wavelength. The metal NPs can also effectively couple thermal and light energy sources to more efficiently drive chemical transformations.

An effective energy boost: Electrons in nonplasmonic transition-metal nanoparticles absorb light energy by interband absorption (see picture) and drive a wide range of well-established organic reactions with high efficiency at ambient temperatures.
07 Feb 13:17
by Ankur Gupta, Thijs J. Aartsma and Gerard W. Canters

Journal of the American Chemical Society
DOI: 10.1021/ja411078b

![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) C coupling (doublet→quartet) and hydrogen atom transfer (quartet→sextet) steps. The carbon atom in MoC+ plays a key role in methane activation because it becomes sp3 hybridized in the initial stages of the ethylene elimination reaction, which leads to much lower energy barriers and more stable intermediates. This study provides insights into the C
C coupling (doublet→quartet) and hydrogen atom transfer (quartet→sextet) steps. The carbon atom in MoC+ plays a key role in methane activation because it becomes sp3 hybridized in the initial stages of the ethylene elimination reaction, which leads to much lower energy barriers and more stable intermediates. This study provides insights into the C