Hyeshik Chang
Shared posts
Evaluating bias-reducing protocols for RNA sequencing library preparation
Translational profiling of clock cells reveals circadianly synchronized protein synthesis.
  |
Related Articles |
Translational profiling of clock cells reveals circadianly synchronized protein synthesis.
PLoS Biol. 2013 Nov;11(11):e1001703
Authors: Huang Y, Ainsley JA, Reijmers LG, Jackson FR
Abstract
Genome-wide studies of circadian transcription or mRNA translation have been hindered by the presence of heterogeneous cell populations in complex tissues such as the nervous system. We describe here the use of a Drosophila cell-specific translational profiling approach to document the rhythmic "translatome" of neural clock cells for the first time in any organism. Unexpectedly, translation of most clock-regulated transcripts--as assayed by mRNA ribosome association--occurs at one of two predominant circadian phases, midday or mid-night, times of behavioral quiescence; mRNAs encoding similar cellular functions are translated at the same time of day. Our analysis also indicates that fundamental cellular processes--metabolism, energy production, redox state (e.g., the thioredoxin system), cell growth, signaling and others--are rhythmically modulated within clock cells via synchronized protein synthesis. Our approach is validated by the identification of mRNAs known to exhibit circadian changes in abundance and the discovery of hundreds of novel mRNAs that show translational rhythms. This includes Tdc2, encoding a neurotransmitter synthetic enzyme, which we demonstrate is required within clock neurons for normal circadian locomotor activity.
PMID: 24348200 [PubMed - indexed for MEDLINE]
Gene set analysis methods: statistical models and methodological differences
Many methods of gene set analysis developed in recent years have been compared empirically in a number of comprehensive review articles. Although it is recognized that different methods tend to identify different gene sets as significant, no consensus has been worked out as to which method is preferable, as the recommendations are often contradictory. In this article, we want to group and compare different methods in terms of the methodological assumptions pertaining to definition of a sample and formulation of the actual null hypothesis. We discuss four models of statistical experiment explicitly or implicitly assumed by most if not all currently available methods of gene set analysis. We analyse validity of the models in the context of the actual biological experiment. Based on this, we recommend a group of methods that provide biologically interpretable results in statistically sound way. Finally, we demonstrate how correlated or low signal-to-noise data affects performance of different methods, observed in terms of the false-positive rate and power.
'Bad fat' may be good for cancer patients
Codon-by-Codon Modulation of Translational Speed and Accuracy Via mRNA Folding
by Jian-Rong Yang, Xiaoshu Chen, Jianzhi Zhang
Rapid cell growth demands fast protein translational elongation to alleviate ribosome shortage. However, speedy elongation undermines translational accuracy because of a mechanistic tradeoff. Here we provide genomic evidence in budding yeast and mouse embryonic stem cells that the efficiency–accuracy conflict is alleviated by slowing down the elongation at structurally or functionally important residues to ensure their translational accuracies while sacrificing the accuracy for speed at other residues. Our computational analysis in yeast with codon resolution suggests that mRNA secondary structures serve as elongation brakes to control the speed and hence the fidelity of protein translation. The position-specific effect of mRNA folding on translational accuracy is further demonstrated experimentally by swapping synonymous codons in a yeast transgene. Our findings explain why highly expressed genes tend to have strong mRNA folding, slow translational elongation, and conserved protein sequences. The exquisite codon-by-codon translational modulation uncovered here is a testament to the power of natural selection in mitigating efficiency–accuracy conflicts, which are prevalent in biology.Derivation and characterization of Dicer- and microRNA-deficient human cells.
|
|
Related Articles |
Derivation and characterization of Dicer- and microRNA-deficient human cells.
RNA. 2014 Jun;20(6):923-37
Authors: Bogerd HP, Whisnant AW, Kennedy EM, Flores O, Cullen BR
Abstract
We have used genome editing to generate inactivating deletion mutations in all three copies of the dicer (hdcr) gene present in the human cell line 293T. As previously shown in murine ES cells lacking Dicer function, hDcr-deficient 293T cells are severely impaired for the production of mature microRNAs (miRNAs). Nevertheless, RNA-induced silencing complexes (RISCs) present in these hDcr-deficient cells are readily programmed by transfected, synthetic miRNA duplexes to repress mRNAs bearing either fully or partially complementary targets, including targets bearing incomplete seed homology to the introduced miRNA. Using these hDcr-deficient 293T cells, we demonstrate that human pre-miRNA processing can be effectively rescued by ectopic expression of the Drosophila Dicer 1 protein, but only in the presence of the PB isoform of Loquacious (Loqs-PB), the fly homolog of the hDcr cofactor TRBP. In contrast, Drosophila Dicer 2, even in the presence of its cofactors Loqs-PD and R2D2, was unable to support human pre-miRNA processing. Interestingly, although ectopic Drosophila Dicer 1/Loqs-PB or hDcr both rescued pre-miRNA processing effectively in these hDcr-deficient cells, there were significant differences in the ratio of the miRNA isoforms that were produced, especially in the case of miR-30 family members, and we also noted differences in the relative expression level of miRNAs vs. passenger strands for a subset of human miRNAs. These data demonstrate that the mechanisms underlying the accurate processing of pre-miRNAs are largely, but not entirely, conserved between mammalian and insect cells.
PMID: 24757167 [PubMed - indexed for MEDLINE]
The role of RNA structure at 5' untranslated region in microRNA-mediated gene regulation [BIOINFORMATICS]
A systematic survey shows increased levels of secondary structure in the 5' UTR near the 5' cap in mRNAs that are regulated by miRNA in animals, but not in plants.
Healing for destruction: tRNA intron degradation in yeast is a two-step cytoplasmic process catalyzed by tRNA ligase Rlg1 and 5'-to-3' exonuclease Xrn1 [Research Communications]
In eukaryotes and archaea, tRNA splicing generates free intron molecules. Although ~600,000 introns are produced per generation in yeast, they are barely detectable in cells, indicating efficient turnover of introns. Through a genome-wide search for genes involved in tRNA biology in yeast, we uncovered the mechanism for intron turnover. This process requires healing of the 5' termini of linear introns by the tRNA ligase Rlg1 and destruction by the cytoplasmic tRNA quality control 5'-to-3' exonuclease Xrn1, which has specificity for RNAs with 5' monophosphate.
Lariat intronic RNAs in the cytoplasm of Xenopus tropicalis oocytes [ARTICLE]
Deep sequencing of cytoplasmic RNA from the oocyte of the frog Xenopus reveals a population of stable lariat molecules derived from specific introns of several thousand protein-coding genes. The existence of stable intronic sequence (sis) RNAs in the cytoplasm of the oocyte, their transmission to the fertilized egg, and their persistence during early embryogenesis suggest that they might play a role in development, perhaps in regulating mRNA translation.
Contribution of calumin to embryogenesis through participation in the endoplasmic reticulum-associated degradation activity
Source:Developmental Biology, Volume 393, Issue 1
Author(s): Shinichiro Yamamoto , Tetsuo Yamazaki , Shinji Komazaki , Takeshi Yamashita , Masako Osaki , Masaya Matsubayashi , Hiroyasu Kidoya , Nobuyuki Takakura , Daiju Yamazaki , Sho Kakizawa
Calumin is an endoplasmic reticulum (ER)-transmembrane protein, and little is known about its physiological roles. Here we showed that calumin homozygous mutant embryos die at embryonic days (E) 10.5−11.5. At mid-gestation, calumin was expressed predominantly in the yolk sac. Apoptosis was enhanced in calumin homozygous mutant yolk sacs at E9.5, pointing to a possible link to the embryonic lethality. Calumin co-immunoprecipitated with ERAD components such as p97, BIP, derlin-1, derlin-2 and VIMP, suggesting its involvement in ERAD. Indeed, calumin knockdown in HEK 293 cells resulted in ERAD being less efficient, as demonstrated by attenuation in both degradations of a misfolded α1-antitrypsin variant and the ER-to-cytosol dislocation of cholera toxin A1 subunit. In calumin homozygous mutant yolk sac endoderm cells, ER stress-associated alterations were observed, including lipid droplet accumulation, fragmentation of the ER and dissociation of ribosomes from the ER. In this context, the ER-overload response, assumed to be cytoprotective, was also triggered in the mutant endoderm cells, but seemed to fully counteract the excessive ER stress generated due to defective ERAD. Taken together, our findings suggested that calumin serves to maintain the yolk sac integrity through participation in the ERAD activity, contributing to embryonic development.
Microtubule Self-Organization via Protein-RNA Network Crosstalk
Source:Cell, Volume 158, Issue 2
Author(s): Charlotte H. Coles , Frank Bradke
Microtubule plus-end tracking proteins are crucial for the regulation of microtubule dynamics. Preitner et al. report that one such protein, adenomatous polyposis coli (APC), also binds RNA and identify mRNAs encoding tubulin subunits within the brain APC-RNA interactome, suggesting a new mode of microtubule self-regulation.
Teaser
Microtubule plus-end tracking proteins are crucial for the regulation of microtubule dynamics. Preitner et al. report that one such protein, adenomatous polyposis coli (APC), also binds RNA and identify mRNAs encoding tubulin subunits within the brain APC-RNA interactome, suggesting a new mode of microtubule self-regulation.APC Is an RNA-Binding Protein, and Its Interactome Provides a Link to Neural Development and Microtubule Assembly
Source:Cell, Volume 158, Issue 2
Author(s): Nicolas Preitner , Jie Quan , Dan W. Nowakowski , Melissa L. Hancock , Jianhua Shi , Joseph Tcherkezian , Tracy L. Young-Pearse , John G. Flanagan
Adenomatous polyposis coli (APC) is a microtubule plus-end scaffolding protein important in biology and disease. APC is implicated in RNA localization, although the mechanisms and functional significance remain unclear. We show APC is an RNA-binding protein and identify an RNA interactome by HITS-CLIP. Targets were highly enriched for APC-related functions, including microtubule organization, cell motility, cancer, and neurologic disease. Among the targets is β2B-tubulin, known to be required in human neuron and axon migration. We show β2B-tubulin is synthesized in axons and localizes preferentially to dynamic microtubules in the growth cone periphery. APC binds the β2B-tubulin 3′ UTR; experiments interfering with this interaction reduced β2B-tubulin mRNA axonal localization and expression, depleted dynamic microtubules and the growth cone periphery, and impaired neuron migration. These results identify APC as a platform binding functionally related protein and RNA networks, and suggest a self-organizing model for the microtubule to localize synthesis of its own subunits.
Graphical abstract

Teaser
APC is an RNA-binding protein whose mRNA interactome is highly enriched for APC-related functions, suggesting coordinated translation and complex assembly. Interaction between APC and β2B-tubulin mRNA is critical for axonal dynamic microtubule extension and neuron migration.RNA Binding of PRC2: Promiscuous or Well Ordered?
Source:Molecular Cell, Volume 55, Issue 2
Author(s): Markus Kretz , Gunter Meister
The polycomb repressive complex 2 (PRC2) methylates histones for epigenetic silencing and associates with thousands of protein-coding and noncoding RNAs. How the recruitment of PRC2 to specific sites is facilitated is currently unclear. Two recent studies have deciphered the impact of RNA binding on PRC2 recruitment and activity (Cifuentes-Rojas et al., 2014; Davidovich et al., 2013).
Teaser
The polycomb repressive complex 2 (PRC2) methylates histones for epigenetic silencing and associates with thousands of protein-coding and noncoding RNAs. How the recruitment of PRC2 to specific sites is facilitated is currently unclear. Two recent studies have deciphered the impact of RNA binding on PRC2 recruitment and activity.Dynamic Heterogeneity and DNA Methylation in Embryonic Stem Cells
Source:Molecular Cell, Volume 55, Issue 2
Author(s): Zakary S. Singer , John Yong , Julia Tischler , Jamie A. Hackett , Alphan Altinok , M. Azim Surani , Long Cai , Michael B. Elowitz
Cell populations can be strikingly heterogeneous, composed of multiple cellular states, each exhibiting stochastic noise in its gene expression. A major challenge is to disentangle these two types of variability and to understand the dynamic processes and mechanisms that control them. Embryonic stem cells (ESCs) provide an ideal model system to address this issue because they exhibit heterogeneous and dynamic expression of functionally important regulatory factors. We analyzed gene expression in individual ESCs using single-molecule RNA-FISH and quantitative time-lapse movies. These data discriminated stochastic switching between two coherent (correlated) gene expression states and burst-like transcriptional noise. We further showed that the “2i” signaling pathway inhibitors modulate both types of variation. Finally, we found that DNA methylation plays a key role in maintaining these metastable states. Together, these results show how ESC gene expression states and dynamics arise from a combination of intrinsic noise, coherent cellular states, and epigenetic regulation.
Graphical abstract

Teaser
Using single-molecule RNA-FISH and quantitative time-lapse movies, Singer et al. discriminate two primary sources of gene expression heterogeneity in embryonic stem cells: random switching between correlated gene expression states and burst-like transcriptional noise. Perturbing specific signaling pathways modulates both processes, and DNA methylation is key to maintaining these states.P granules
Source:Current Biology, Volume 24, Issue 14
Author(s): Jennifer T. Wang , Geraldine Seydoux
Teaser
Wang and Seydoux discuss the functional importance of P granules – the germline-specific RNA granules of C. elegans.Spotlight falls on top 1% in science
Spotlight falls on top 1% in science
Nature 511, 7510 (2014). doi:10.1038/511387e
Author: Chris Woolston
Nature's roundup of the papers and issues gaining traction on social media.News that a rarified group of scientists has claimed the lion's share of publications has set off a social-media discussion about the fairness of the system. Researchers also took to Twitter to
Elite male biologists hire fewer women [Social Sciences]
Peer review, reviewed

Revise and resubmit.
Rebecca Schuman, who has almost single-handedly turned Slate into one of best big websites for coverage of the many trials and tribulations of academia, turns to peer review for scholarly journals, in which an author’s academic peers volunteer to weigh in on whether a manuscript is worthy of publication. Schuman discusses the problems of of both how long the process takes—routinely more than a year, especially with the back-and-forth of revisions—and tone:
Think of your meanest high school mean girl at her most gleefully, underminingly vicious. Now give her a doctorate in your discipline, and a modicum of power over your future. That’s peer review.
And she suggests something that might sound familiar to those of us who hang out in the evolutionary ecology blog-o-verse: enforced reviewing reciprocity.
… what if in order to be eligible to submit an academic article to a journal, a scholar had first to volunteer to review someone else’s article for that same journal? … You want to publish and not perish? First you have to earn that right by making a punctual, non-petty investment into the publishing enterprise.
Paging Jeremy Fox:
A few years ago, Owen Petchey and I proposed a reform known as PubCreds, the purpose of which was to oblige authors to review in appropriate proportion to how much they submit. For instance, if each paper you submit receives two reviews, then arguably you ought to perform two reviews for every paper you submit.
Fox and Petchey and Lindsay Haddan actually tested the hypothesis that the average ecologist receives more reviewing services than he provides, using data on papers sent out for review by the journals of the British Ecological Society. They found (given caveats and follow-up discussion in the post linked above) that the relationship between reviewing and being reviewed was actually pretty well balanced. While a number of authors contributed fewer reviews than their own papers required, another large group reviewed more than required.
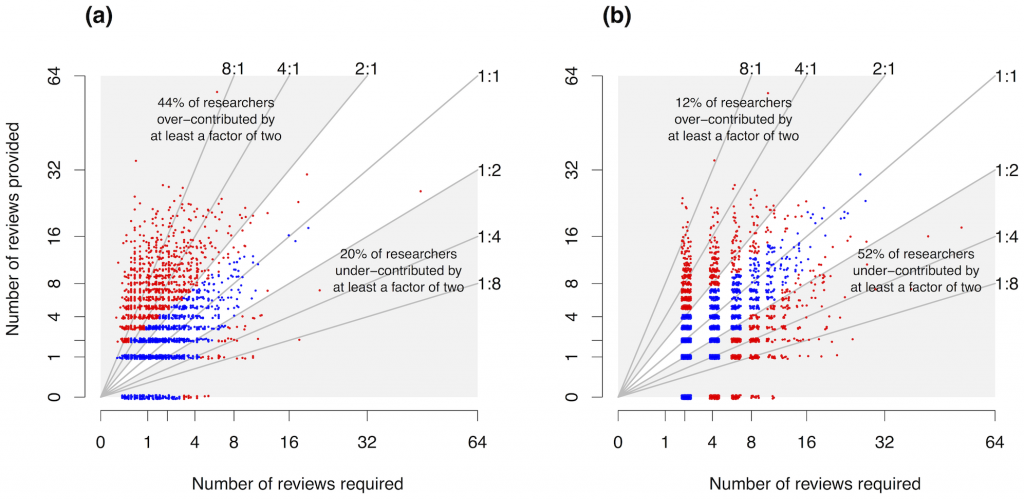
Depending on different assumptions about the relationship between reviewing and the need for review, from 12 to 44% of authors for BES journals did at least twice as much reviewing as they “needed” to; and from 20 to 52% did less than half. Figure 1 from Petchey et al. (2014).
Of course, things may be quite different in the humanities, which are Schuman’s academic homeland. That may also explain why Schuman doesn’t bring up the other problem of peer review that occupies most scientists, of finding a journal at the right “level” for a paper—i.e., determining whether the paper is novel or important enough to match a particular journal’s (self-image of) prestige. In my experience, which has mostly involved faster reviewing time frames than Schuman describes, the real time-suck in publishing comes when you submit to a prestigious journal, go through review, and end up with a decision along the lines of “this is nice, but it’s not a big enough deal for us.” Then you have to reformat, and often extensively re-write, for a different venue a little farther down the prestige ladder. Review at each journal might be timely and constructive, but by the time the paper appears in print you’ve burned 18 months.
This problem is the motivation for the Axios pre-submission review service (co-managed by our own Tim Vines), in which authors submit manuscripts with a list of possible target journals, and reviewers select the best match—then forward their reviews and the manuscript, revised accordingly, to that journal. It’s a workaround, but as the number of participating journals increases, it promises to substantially reduce the time to publication.
Apart from the prestige-level-finding problem, I have generally had pretty good peer review experiences—and in the one or two cases where a reviewer behaved badly, the journal’s editor treated the bad review appropriately. (There is, of course, the caveat that any peer review process that ends with a published paper feels like it went all right in the end.) Everyone would like reviews to happen faster, but I think most of us put them on the calendar just before the official deadline, and it’s never difficult to think of other things on one’s to-do list that should happen before writing a review.
As for the differences between science and the humanities—I am currently awaiting reviews of my first truly interdisciplinary manuscript. Maybe I’ll feel quite differently in a week or two, or ten.
Reference
Petchey O.L. & Lindsay Haddon (2014). Imbalance in individual researcher’s peer review activities quantified for four British Ecological Society journals, 2003-2010, PLoS ONE, 9 (3) e92896. DOI: http://dx.doi.org/10.1371/journal.pone.0092896
Regulation of histone mRNA SLBP by phosphorylation [Biochemistry]
PRADA: pipeline for RNA sequencing data analysis
Summary: Technological advances in high-throughput sequencing necessitate improved computational tools for processing and analyzing large-scale datasets in a systematic automated manner. For that purpose, we have developed PRADA (Pipeline for RNA-Sequencing Data Analysis), a flexible, modular and highly scalable software platform that provides many different types of information available by multifaceted analysis starting from raw paired-end RNA-seq data: gene expression levels, quality metrics, detection of unsupervised and supervised fusion transcripts, detection of intragenic fusion variants, homology scores and fusion frame classification. PRADA uses a dual-mapping strategy that increases sensitivity and refines the analytical endpoints. PRADA has been used extensively and successfully in the glioblastoma and renal clear cell projects of The Cancer Genome Atlas program.
Availability and implementation: http://sourceforge.net/projects/prada/
Contact: gadgetz@broadinstitute.org or rverhaak@mdanderson.org
Supplementary information: Supplementary data are available at Bioinformatics online.
Computational identification of natural peptides based on analysis of molecular evolution
Motivation: Many secretory peptides are synthesized as inactive precursors that must undergo post-translational processing to become biologically active peptides. Attempts to predict natural peptides are limited by the low performance of proteolytic site predictors and by the high combinatorial complexity of pairing such sites. To overcome these limitations, we analyzed the site-wise evolutionary mutation rates of peptide hormone precursors, calculated using the Rate4Site algorithm.
Results: Our analysis revealed that within their precursors, peptide residues are significantly more conserved than the pro-peptide residues. This disparity enables the prediction of peptides with a precision of ~60% at a recall of 40% [receiver-operating characteristic curve (ROC) AUC 0.79]. Subsequently, combining the Rate4Site score with additional features and training a Random Forest classifier enable the prediction of natural peptides hidden within secreted human proteins at a precision of ~90% at a recall of 50% (ROC AUC 0.96). The high performance of our method allows it to be applied to full secretomes and to predict naturally occurring active peptides. Our prediction on Homo sapiens revealed several putative peptides in the human secretome that are currently unannotated. Furthermore, the unique expression of some of these peptides implies a potential hormone function, including peptides that are highly expressed in endocrine glands.
Availability and implementation: A pseudocode is available in the Supplementary information.
Contact: doron.gerber@biu.ac.il or kliger@cgen.com
Supplementary information: Supplementary data are available at Bioinformatics online.
Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway
Nature advance online publication 09 July 2014. doi:10.1038/nature13429
Authors: Ashton Trey Belew, Arturas Meskauskas, Sharmishtha Musalgaonkar, Vivek M. Advani, Sergey O. Sulima, Wojciech K. Kasprzak, Bruce A. Shapiro & Jonathan D. Dinman
Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins
Nature advance online publication 09 July 2014. doi:10.1038/nature13466
Authors: Hani Goodarzi, Steven Zhang, Colin G. Buss, Lisa Fish, Saeed Tavazoie & Sohail F. Tavazoie
Aberrant regulation of RNA stability has an important role in many disease states. Deregulated post-transcriptional modulation, such as that governed by microRNAs targeting linear sequence elements in messenger RNAs, has been implicated in the progression of many cancer types. A defining feature of RNA is its ability to fold into structures. However, the roles of structural mRNA elements in cancer progression remain unexplored. Here we performed an unbiased search for post-transcriptional modulators of mRNA stability in breast cancer by conducting whole-genome transcript stability measurements in poorly and highly metastatic isogenic human breast cancer lines. Using a computational framework that searches RNA sequence and structure space, we discovered a family of GC-rich structural cis-regulatory RNA elements, termed sRSEs for structural RNA stability elements, which are significantly overrepresented in transcripts displaying reduced stability in highly metastatic cells. By integrating computational and biochemical approaches, we identified TARBP2, a double-stranded RNA-binding protein implicated in microRNA processing, as the trans factor that binds the sRSE family and similar structural elements—collectively termed TARBP2-binding structural elements (TBSEs)—in transcripts. TARBP2 is overexpressed in metastatic cells and metastatic human breast tumours and destabilizes transcripts containing TBSEs. Endogenous TARBP2 promotes metastatic cell invasion and colonization by destabilizing amyloid precursor protein (APP) and ZNF395 transcripts, two genes previously associated with Alzheimer’s and Huntington’s disease, respectively. We reveal these genes to be novel metastasis suppressor genes in breast cancer. The cleavage product of APP, extracellular amyloid-α peptide, directly suppresses invasion while ZNF395 transcriptionally represses a pro-metastatic gene expression program. The expression levels of TARBP2, APP and ZNF395 in human breast carcinomas support their experimentally uncovered roles in metastasis. Our findings establish a non-canonical and direct role for TARBP2 in mammalian gene expression regulation and reveal that regulated RNA destabilization through protein-mediated binding of mRNA structural elements can govern cancer progression.
DNA methylation dynamics of the human preimplantation embryo
Nature advance online publication 23 July 2014. doi:10.1038/nature13581
Authors: Zachary D. Smith, Michelle M. Chan, Kathryn C. Humm, Rahul Karnik, Shila Mekhoubad, Aviv Regev, Kevin Eggan & Alexander Meissner
In mammals, cytosine methylation is predominantly restricted to CpG dinucleotides and stably distributed across the genome, with local, cell-type-specific regulation directed by DNA binding factors. This comparatively static landscape is in marked contrast with the events of fertilization, during which the paternal genome is globally reprogrammed. Paternal genome demethylation includes the majority of CpGs, although methylation remains detectable at several notable features. These dynamics have been extensively characterized in the mouse, with only limited observations available in other mammals, and direct measurements are required to understand the extent to which early embryonic landscapes are conserved. We present genome-scale DNA methylation maps of human preimplantation development and embryonic stem cell derivation, confirming a transient state of global hypomethylation that includes most CpGs, while sites of residual maintenance are primarily restricted to gene bodies. Although most features share similar dynamics to those in mouse, maternally contributed methylation is divergently targeted to species-specific sets of CpG island promoters that extend beyond known imprint control regions. Retrotransposon regulation is also highly diverse, and transitions from maternally to embryonically expressed elements. Together, our data confirm that paternal genome demethylation is a general attribute of early mammalian development that is characterized by distinct modes of epigenetic regulation.
The DNA methylation landscape of human early embryos
Nature advance online publication 23 July 2014. doi:10.1038/nature13544
Authors: Hongshan Guo, Ping Zhu, Liying Yan, Rong Li, Boqiang Hu, Ying Lian, Jie Yan, Xiulian Ren, Shengli Lin, Junsheng Li, Xiaohu Jin, Xiaodan Shi, Ping Liu, Xiaoye Wang, Wei Wang, Yuan Wei, Xianlong Li, Fan Guo, Xinglong Wu, Xiaoying Fan, Jun Yong, Lu Wen, Sunney X. Xie, Fuchou Tang & Jie Qiao
DNA methylation is a crucial element in the epigenetic regulation of mammalian embryonic development. However, its dynamic patterns have not been analysed at the genome scale in human pre-implantation embryos due to technical difficulties and the scarcity of required materials. Here we systematically profile the methylome of human early embryos from the zygotic stage through to post-implantation by reduced representation bisulphite sequencing and whole-genome bisulphite sequencing. We show that the major wave of genome-wide demethylation is complete at the 2-cell stage, contrary to previous observations in mice. Moreover, the demethylation of the paternal genome is much faster than that of the maternal genome, and by the end of the zygotic stage the genome-wide methylation level in male pronuclei is already lower than that in female pronuclei. The inverse correlation between promoter methylation and gene expression gradually strengthens during early embryonic development, reaching its peak at the post-implantation stage. Furthermore, we show that active genes, with the trimethylation of histone H3 at lysine 4 (H3K4me3) mark at the promoter regions in pluripotent human embryonic stem cells, are essentially devoid of DNA methylation in both mature gametes and throughout pre-implantation development. Finally, we also show that long interspersed nuclear elements or short interspersed nuclear elements that are evolutionarily young are demethylated to a milder extent compared to older elements in the same family and have higher abundance of transcripts, indicating that early embryos tend to retain higher residual methylation at the evolutionarily younger and more active transposable elements. Our work provides insights into the critical features of the methylome of human early embryos, as well as its functional relation to the regulation of gene expression and the repression of transposable elements.
Regulation of microRNA biogenesis
Nature Reviews Molecular Cell Biology 15, 509 (2014). doi:10.1038/nrm3838
Authors: Minju Ha & V. Narry Kim
MicroRNAs (miRNAs) are small non-coding RNAs that function as guide molecules in RNA silencing. Targeting most protein-coding transcripts, miRNAs are involved in nearly all developmental and pathological processes in animals. The biogenesis of miRNAs is under tight temporal and spatial control, and their dysregulation is
Small RNAs break out: the molecular cell biology of mobile small RNAs
Nature Reviews Molecular Cell Biology 15, 525 (2014). doi:10.1038/nrm3840
Authors: Peter Sarkies & Eric A. Miska
Small RNAs that function in a non-cell autonomous manner are becoming increasingly recognized as regulatory molecules with the potential to transmit information between cells, organisms and species. In plants and nematodes, small RNA mobility can be genetically dissected to provide information about the nature of
Identification and consequences of miRNA–target interactions — beyond repression of gene expression
Improving biotech education through gamified laboratory simulations
Nature Biotechnology 32, 694 (2014). doi:10.1038/nbt.2955
Authors: Mads T Bonde, Guido Makransky, Jakob Wandall, Mette V Larsen, Mikkel Morsing, Hanne Jarmer & Morten O A Sommer
Gamified laboratory simulations motivate students and improve learning outcomes compared with traditional teaching methods.
High-efficiency translational bypassing of non-coding nucleotides specified by mRNA structure and nascent peptide
Article
Translational bypassing occurs when the ribosome skips nucleotides downstream of a take-off codon and re-initiates protein synthesis at a location further down on the mRNA. Here, Samatova et al. show that translational bypassing of bacteriophage T4 gene60 mRNA is specified both by the mRNA sequence and the nascent peptide.
Nature Communications doi: 10.1038/ncomms5459
Authors: Ekaterina Samatova, Andrey L. Konevega, Norma M. Wills, John F. Atkins, Marina V. Rodnina