04 Jan 16:18
by Vasudev R. Bhonde, Brian T. O'Neill, Stephen L. Buchwald
Abstract
The development of a palladacyclic precatalyst supported by a new biaryl(dialkyl)phosphine ligand (VPhos) in combination with octanoic acid/sodium octanoate as a simple and effective surfactant system provided an improved catalyst system for the rapid construction of a broad spectrum of alkylated scaffolds from alkyl zinc reagents generated in situ.

Happy to get its feet wet: VPhos was developed as a hybrid ligand incorporating structural elements of existing ligands for improved activity in the micelle-enhanced palladium-catalyzed cross-coupling of non-aromatic O- and N-heterocyclic alkyl bromides with (hetero)aryl halides (see scheme). The Pd/VPhos catalyst (5 mol %) and a simple surfactant system based on octanoic acid enabled the efficient synthesis of a broad range of alkylated (hetero)arenes.
07 Dec 14:20
by Yuki Mizukami, Zhiyi Song and Tamotsu Takahashi

Organic Letters
DOI: 10.1021/acs.orglett.5b02589

07 Dec 14:05
by Jian Zheng, Jin-Hong Lin, Liu-Ying Yu, Yun Wei, Xing Zheng and Ji-Chang Xiao

Organic Letters
DOI: 10.1021/acs.orglett.5b03159

30 Nov 14:47
by Sina Rösler, Michael Ertl, Torsten Irrgang, Rhett Kempe

Amine Alkylation In their Communication on page 15046 ff., R. Kempe et al. describe the alkylation of amines by alcohols in the presence of a Co catalyst under mild conditions. Selectively monoalkylated aromatic amines as well as unsymmetrically substituted diamines are obtained.
21 Nov 19:35
by Maximilian Joost, Abderrahmane Amgoune, Didier Bourissou
Abstract
For a while, the reactivity of gold complexes was largely dominated by their Lewis acid behavior. In contrast to the other transition metals, the elementary steps of organometallic chemistry—oxidative addition, reductive elimination, transmetallation, migratory insertion—have scarcely been studied in the case of gold or even remained unprecedented until recently. However, within the last few years, the ability of gold complexes to undergo these fundamental reactions has been unambiguously demonstrated, and the reactivity of gold complexes was shown to extend well beyond π-activation. In this Review, the main achievements described in this area are presented in a historical context. Particular emphasis is set on mechanistic studies and structure determination of key intermediates. The electronic and structural parameters delineating the reactivity of gold complexes are discussed, as well as the remaining challenges.

Sitting on a gold mine? In the past few years, the reactivity of gold complexes has been extended well beyond Lewis acid behavior and electrophilic activation of π-substrates. Elementary steps considered highly unlikely, if not impossible, for gold complexes have been unambiguously demonstrated, offering new perspectives in gold catalysis.
21 Nov 18:59
by Joydev K. Laha, Krupal P. Jethava and Sagarkumar Patel

Organic Letters
DOI: 10.1021/acs.orglett.5b03071

11 Nov 10:31
Chem. Sci., 2016, 7,85-88
DOI: 10.1039/C5SC03025K, Edge Article

Open Access
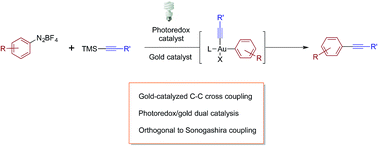
Suhong Kim, Jaime Rojas-Martin, F. Dean Toste
A new method for the alkynylation of aryldiazonium salts with TMS-alkynes via dual gold and photoredox catalysis is described.
The content of this RSS Feed (c) The Royal Society of Chemistry

10 Nov 09:40
by Marius Hervé, Guillaume Lefèvre, Emily A. Mitchell, Bert U. W. Maes, Anny Jutand
Abstract
The mechanism of Stille reactions (cross-coupling of ArX with Ar′SnnBu3) performed in the presence of fluoride ions is established. A triple role for fluoride ions is identified from kinetic data on the rate of the reactions of trans-[ArPdBr(PPh3)2] (Ar=Ph, p-(CN)C6H4) with Ar′SnBu3 (Ar′=2-thiophenyl) in the presence of fluoride ions. Fluoride ions promote the rate-determining transmetallation by formation of trans-[ArPdF(PPh3)2], which reacts with Ar′SnBu3 (Ar′=Ph, 2-thiophenyl) at room temperature, in contrast to trans-[ArPdBr(PPh3)2], which is unreactive. However, the concentration ratio [F−]/[Ar′SnBu3] must not be too high, because of the formation of unreactive anionic stannate [Ar′Sn(F)Bu3]−. This rationalises the two kinetically antagonistic roles exerted by the fluoride ions that are observed experimentally, and is found to be in agreement with the kinetic law. In addition, fluoride ions promote reductive elimination from trans-[ArPdAr′(PPh3)2] generated in the transmetallation step.

Fluoride does the job: Three roles for fluoride ions in Stille reactions are identified from kinetic data. F− promotes the rate-determining transmetallation by formation of trans-[ArPdF(PPh3)2], which reacts with Ar′SnBu3, in contrast to trans-[ArPdBr(PPh3)2], which does not. F− also promotes the reductive elimination in trans-[ArPdAr′(PPh3)2]. However, the concentration ratio [F−]/[Ar′SnBu3] must be less than 1 because of the formation of unreactive [Ar′Sn(F)Bu3]−.
09 Nov 09:26
by Wenyong Han, Xiaojian Zhou, Siyi Yang, Guangyan Xiang, Baodong Cui and Yongzheng Chen

The Journal of Organic Chemistry
DOI: 10.1021/acs.joc.5b02145

04 Nov 11:53
by Takashi Niwa, Hidenori Ochiai, Yasuyoshi Watanabe and Takamitsu Hosoya

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b10119

02 Nov 11:50
by Fatima Rebih, Manuel Andreini, Aurélien Moncomble, Anne Harrison-Marchand, Jacques Maddaluno, Muriel Durandetti
Abstract
A novel Ni0-catalyzed carboxylation of aryl tosylates with carbon dioxide has been achieved under moderate temperatures and atmospheric pressure. In this procedure, the active Ni0 species is generated in situ by simply mixing the Ni0 precatalyst [NiBr2(bipy)] with an excess of manganese metal. This approach requires neither a glove-box nor the tedious preparation of sophisticated intermediate organometallic derivatives. This mild, convenient, and user-friendly process is successfully applied to the valorization of carbon dioxide and the synthesis of versatile reactants with broad tolerance of substituents.

Direct carboxylation of aryl tosylates by CO2 is rendered possible by a Ni0 catalyst, generated in situ by reduction of [NiBr2(bipy)] with manganese metal. This approach, which requires neither a glove-box nor the preparation of a sensitive organometallic derivative, is applicable to a wide range of aromatic phenols, bearing either electron-donating or withdrawing substituents.
29 Oct 13:39
by XiaoZhi Lim
How to make the most of carbon dioxide
Nature 526, 7575 (2015). http://www.nature.com/doifinder/10.1038/526628a
Author: XiaoZhi Lim
Researchers hope to show that using the gas as a raw material could make an impact on climate change.

29 Oct 13:36
by Jiawei Rong, Tilde Pellegrini, Syuzanna R. Harutyunyan
Abstract
Catalytic enantioselective addition of organometallic nucleophiles to ketones is among the most straightforward approaches to the synthesis of chiral tertiary alcohols. The first such catalytic methodologies using the highly reactive organomagnesium reagents, which are the preferred organometallic reagents in terms of cost, availability, atom efficiency, and structural diversity, were developed only during the last five years. This Concept article highlights the fundamental breakthrough that made the development of methodologies for highly enantioselective CuI-catalyzed alkylation of ketones using organomagnesium reagents possible.

Catalytic enantioselective addition of organometallic nucleophiles to ketones is among the most straightforward approaches to the synthesis of chiral tertiary alcohols. This concept article highlights the fundamental breakthrough that made the development of methodologies for synthesis of tertiary alcohols via enantioselective CuI-catalyzed addition of organomagnesium reagents to ketones possible.
27 Oct 18:34
by Anna Falk, Alberto Cavalieri, Gary S. Nichol, Dieter Vogt, Hans-Günther Schmalz
Abstract
The asymmetric hydrocyanation of vinylarenes was investigated using hydrogen cyanide (HCN) in the presence of 5 mol% of a catalyst prepared from a phenol-derived chiral phosphine-phosphite ligand and bis(cyclooctadiene)nickel [Ni(cod)2]. The reactions were performed in tetrahydrofuran (THF) at room temperature to give exclusively the branched nitriles with superior enantioselectivities of 88–99% ee for vinylarenes and 74–94% ee for vinylheteroarenes, respectively. Using styrene as a model substrate it was shown that the catalyst loading could be decreased to 0.42 mol% without any loss of selectivity (88% ee). The structure of the pre-catalyst, i.e., a tetrahedral Ni(0)(P,P-chelate)(cod) complex, was proven by X-ray and NMR analysis. Additional insight into the reaction course was gained by monitoring the hydrocyanation of styrene-d8 by means of 2D NMR spectroscopy.

27 Oct 12:31
by Stephan Laue, Verena Haverkamp and Leslaw Mleczko

Organic Process Research & Development
DOI: 10.1021/acs.oprd.5b00183

12 Oct 12:20
by Hyunwoo Kim and Sukbok Chang

ACS Catalysis
DOI: 10.1021/acscatal.5b02165

12 Oct 12:18
by Anna Troiani, Marzio Rosi, Stefania Garzoli, Chiara Salvitti, Giulia de Petris
Abstract
An unusual iron transfer and carbon–carbon coupling take place in gas-phase ionized mixtures containing ferrocene and dichloromethane. Ferrous chloride and the protonated benzenium ion are eventually formed by a thermal and efficient reaction, through stable intermediates that undergo a remarkable reorganization. The mechanism of the concerted iron extrusion, carbon–chlorine bond activation and carbon–carbon bond formation is elucidated by electronic structure calculations that show the crucial role of iron.

Ion–molecule reactions: A remarkable reorganization occured in iron-containing cations under ambient conditions. Fast and effective carbon–chlorine activation and carbon–carbon coupling was observed in gas-phase mixtures containing ferrocene and dichloromethane, through reaction intermediates (see picture) that eventually extrude the iron giving ferrous chloride.
07 Oct 14:55
by Melissa Lee and Melanie S. Sanford

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b09099

07 Oct 07:53
by Daniel T. Weiss, Markus R. Anneser, Stefan Haslinger, Alexander Pöthig, Mirza Cokoja, Jean-Marie Basset and Fritz E. Kühn

Organometallics
DOI: 10.1021/acs.organomet.5b00732

02 Oct 11:04
by Qilin Li, Hongbin Zhang
Abstract
In this paper, a new strategy towards the synthesis of codeine and morphine is reported. This new approach features a cascade cyclization to construct the dihydrofuran ring, and an intramolecular palladium catalyzed C![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) H olefination of unactivated aliphatic alkene to install the morphinan ring system.
H olefination of unactivated aliphatic alkene to install the morphinan ring system.

Milk of the poppy: This paper reports a new strategy towards the synthesis of codeine and morphine, which features a cascade cyclization to construct the dihydrofuran ring and an intramolecular palladium-catalyzed CH olefination of an unactivated aliphatic alkene to install the morphinan ring system.
23 Sep 08:43
by Pan Gao, Wei Guo, Jingjing Xue, Yue Zhao, Yu Yuan, Yuanzhi Xia and Zhuangzhi Shi

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b06758

18 Sep 11:04
Chem. Soc. Rev., 2015, 44,8062-8096
DOI: 10.1039/C5CS00003C, Review Article
Rajeshwer Vanjari, Krishna Nand Singh
This review attempts to describe the latest developments in the utilisation of methylarenes adopting C-H functionalization strategies and covers all the developments until March 2015.
The content of this RSS Feed (c) The Royal Society of Chemistry

18 Sep 09:16
by Takayuki Furukawa, Mamoru Tobisu and Naoto Chatani

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b07677

17 Sep 11:14
by Erica M. D’Amato, Constanze N. Neumann and Tobias Ritter

Organometallics
DOI: 10.1021/acs.organomet.5b00731

15 Sep 07:53
by Nicholas A. Isley, Roscoe T. H. Linstadt, Sean M. Kelly, Fabrice Gallou and Bruce H. Lipshutz

Organic Letters
DOI: 10.1021/acs.orglett.5b02240

14 Sep 10:42
by Matteo Villa, Axel Jacobi von Wangelin

Radical changes: The applicability of alkene hydroamination has recently been significantly expanded by the development of radical variants that are based on initial hydrogen atom transfer to the alkene. This Highlight assesses the current state of the art, focusing on an iron-catalyzed reaction that utilizes stable nitroarenes as the electrophilic N component and is based on the dual catalytic activation of both starting materials.
14 Sep 09:48
by Wei Zhou, Mengyang Fan, Junli Yin, Yongwen Jiang and Dawei Ma

Journal of the American Chemical Society
DOI: 10.1021/jacs.5b08411

11 Sep 08:18
by Carlos Vila, Sara Cembellín, Valentín Hornillos, Massimo Giannerini, Martín Fañanás-Mastral, Ben L. Feringa
Abstract
A Pd-catalyzed direct cross-coupling of two distinct aryl bromides mediated by tBuLi is described. The use of [Pd-PEPPSI-IPr] or [Pd-PEPPSI-IPent] as catalyst allows for the efficient one-pot synthesis of unsymmetrical biaryls at room temperature. The key for this selective cross-coupling is the use of an ortho-substituted bromide that undergoes lithium–halogen exchange preferentially.

Bro to Bro: A palladium-catalyzed direct cross-coupling of two distinct aryl bromides mediated by tBuLi is described. The use of [Pd-PEPPSI-IPr] or [Pd-PEPPSI-IPent] as catalyst allows for the efficient one-pot synthesis of unsymmetrical biaryls at room temperature. The key for this selective cross-coupling is the use of an ortho-substituted bromide that undergoes lithium–halogen exchange preferentially.
11 Sep 08:17
by Jing Zheng, Dahai Wang and Sunliang Cui

Organic Letters
DOI: 10.1021/acs.orglett.5b02294

09 Sep 09:46
by Jacob A. Przyojski, Kevin P. Veggeberg, Hadi D. Arman and Zachary J. Tonzetich

ACS Catalysis
DOI: 10.1021/acscatal.5b01445